

Excipient selection in biologics and vaccines formulation development
Posted: 19 February 2014 | | 1 comment
Excipients are an integral part of pharmaceutical products and play an important role in the formulation development of both small and large molecule pharmaceuticals. The type and extent of excipient use depends on several factors, including the type of active ingredient, route of administration, dosage form, target population and indication and so forth.
With a growing number of protein therapeutics and vaccines in development, stabilisation during processing and storage presents a major challenge for the pharmaceutical industry. Biologics and vaccines are inherently unstable and prone to degradation by several physical and chemical degradation mechanisms. Therefore, a variety of excipients are required to stabilise biologics and vaccines during processing and storage. Selection and use of the appropriate excipients enable development of novel therapies and robust pharmaceutical products. The purpose of this article is to present an overview of the challenges associated with biologic and vaccine formulation development as well as different types of excipients used to stabilise these products.
Excipients
An excipient, by definition, is a pharmacologically inert substance by itself, but when used in combination with an active ingredient it can provide several benefits. Use of excipients in medicinal products date back to medieval times when they were primarily used as a vehicle for active ingredients or flavour to improve the palatability of medicines. In today’s pharmaceutical development, excipients are an integral part of pharmaceutical products, used in multiple ways, and have well defined functional roles. For proteins and vaccines, these roles include (a) enhancing solubility of the active, (b) enhancing process and shelf life stability of the active, (c) controlling pH and tonicity, (d) maintaining preferred stable conformation for proteins or vaccines including exposure of the functional epitopes, (e) preventing aggregation or degradation of the active, (f) several other functions like bulking agents, antioxidants, or preservatives1. Another class of excipients primarily used in vaccines are adjuvants, which are defined as substances that enhance the pharmacological effect of a drug or increase the ability of an antigen to stimulate the immune system2.
Excipients are approved as part of the pharmaceutical products but not alone. They range from well characterised organic or inorganic molecules to more complex and difficult to characterise biological structures. The selection of excipients also depends on the regulatory requirements in the respective markets. In the US, the Food and Drug Administration provides a searchable list of approved inactive ingredients in a database (IID) (www.accessdata.fda.gov/scripts/cder/iig/index.cfm) with their approved concentration, dosage form and route of administration. In Japan, the Japanese Pharmaceutics Excipients Council (JPEC) compiles excipients in approved products, route of administration, and patient exposure in the Japanese Pharmaceutical Excipients Dictionary (JPED). In Canada, Health Canada publishes an acceptable non-medicinal agents list. However, there is no equivalent list or database of excipients approved in drug products in Europe from the European Medicines Agency.
Generally Recognised as Safe (GRAS) excipients
Excipients which originated in the food industry typically have a generally recognised as safe (GRAS) status by the US FDA. The human exposure levels and safety information for excipients designated as GRAS may already be available. It must be noted that even though GRAS excipients are widely screened across vaccines and biologics, the parenteral route of administration limits available excipients. Table 1 provides examples of commonly used excipients in biologics and vaccine formulations.
Novel / new excipients
A novel / new excipient is defined as an excipient which is being used for the first time in a drug product, or by a new route of administration. The excipient may be a new chemical entity or a well-established one that has not yet been used in human products, for the intended administration route or at the proposed level3,4. The International Pharmaceutical Excipients Council (IPEC) is an international industry association that contributes to the development and harmonisation of international excipient standards, the introduction of new excipients and the development of best practice and guidance concerning excipients. Some of the novel excipients being investigated for biologic products are cyclodextrin-based excipients5.
Excipients for biologics and vaccine formulation: Challenges in biologics and vaccine formulation
Biological molecules (peptide, proteins, mAbs and vaccines) are inherently less stable than small molecules and formulation development is often challenging with stringent product development timelines. Additionally, limited availability of drug substance, especially during early development, forces formulation scientists to rely heavily on forced degradation studies using limited analytical techniques. Such approaches often result in suboptimal formulation development, ultimately leading to stability and manufacturing challenges during clinical and commercial manufacturing. Furthermore, complex molecular structure, lack of well-defined stability-indicating assays and a multitude of degradation mechanisms create major hurdles for the formulation development of biologics and vaccines. The understanding of active–excipient interactions is critical in the rational design of formulations to stabilise protein-based therapeutic drugs and vaccines. The complexity of formulation development is often more pronounced for the development of high concentration antibody formulations due to additional constraints of viscosity, analytical characterisation at high concentration and desire to formulate an isotonic formulation while maintaining the ratio of excipient to antibody to provide the stability6.
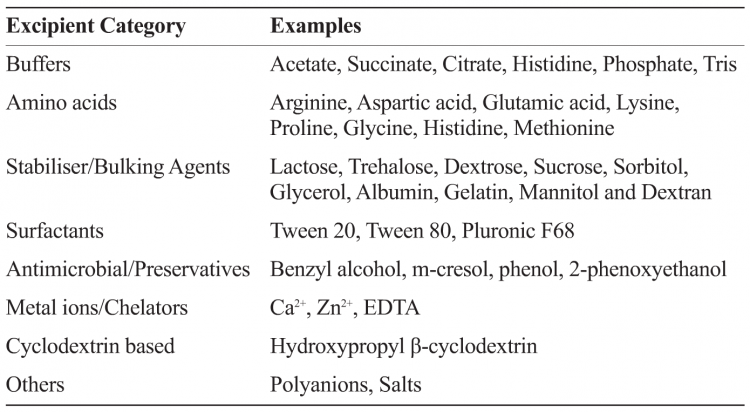
Table 1: Categories and examples of some commonly used pharmaceutical excipients in biologic and vaccine formulations
Formulation development for vaccines poses additional challenges as the vaccine antigen can be of different types, including live attenuated viruses, inactivated viruses, purified recombinant proteins, purified viral proteins, virus-like particles, inactivated bacterial toxins and polysaccharides. Often, multiple antigens are included in a vaccine product in addition to adjuvants. Formulation development for peptide, protein and subunit based antigens can be approached in a similar way to biologics. However, formulation development for attenuated live virus vaccines may require a different approach as the focus is on stabilising the whole viral particles and associated infectivity. Vaccines for emerging markets can benefit from excipients that improve thermostability, especially if it can reduce cold-chain requirements during storage and/or distribution7. Desire for room temperature stability, combination products, multi-dose product and differentiated product with auto-injectors or pre-filled syringes could further add to the challenge. To secure complete alignment between the formulation scientist and the intended market, it is important to develop a global target product profile early in the programme with customer centricity in mind. This will form the basis of the formulation development and ensure successful development of a safe, efficacious, and quality product.
Excipient screening / selection
The formulation development process can, in general, be compartmentalised minimally into three different but interconnected stages, namely pre-formulation, formulation and process development. The pre-formulation stage involves understanding of the biologic on the basis of its amino acid sequence, performing biochemical and biophysical characterisation as a function of pH, ionic strength and excipients to understand the prominent degradation pathways. This stage also involves development of stability-indicating assays, and identification of a few lead excipients using (preferably) a systematic approach such as Design of Experiment (DOE) or Empirical Phase Diagram (EPD)8-10. A comprehensive picture of macromolecular behaviour can be obtained using high-throughput formulation approaches to probe changes in structure (e.g. secondary, tertiary etc.) and function (e.g. activity, potency, binding wherever possible) in the presence of stress condition (e.g. pH, temperature, freeze thaw, drying, sheer). Table 2 provides examples of factors and general ranges tested in formulations screening.
Development of frozen or lyophilised product may be initiated if the initial stability studies suggest that the liquid formulation fails to meet the desired target product profile established early in the programme. Such rational approach to pre-formulation may also provide a basis for employing a QbD (Quality by Design) approach as the programme moves forward through formulation development. Through extensive stability studies (both real-time and accelerated) and in vivo animal model testing (when applicable), a potential formulation must be selected for preclinical and clinical manufacturing. It must be stated that in vivo testing may or may not correlate with human immunogenicity and often serves as a disaster check. Reformulation and/or reprocessing during clinical development may require clinical bridging studies thereby increasing the cost and complexity of the program. It is therefore recommended that process and formulation optimisation occur at an early stage.
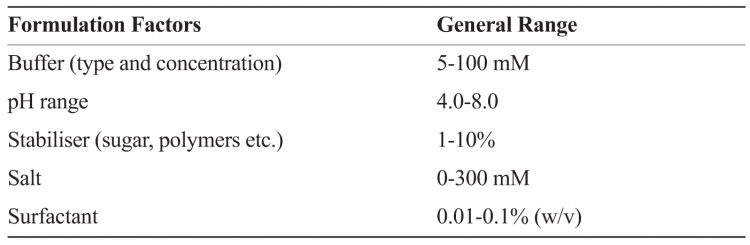
Table 2: Example illustrating factors for determining formulation design space
Liquid formulation development
A mechanistic understanding of the way excipients stabilise biologics is essential for a successful formulation strategy. Forces that impact the conformational stability of the molecule such as electrostatic interactions, Van der Waals interactions, hydrogen bonding and hydrophobicity may be targeted by proper selection of excipients. The native conformation of biologics, for example, can be stabilised by high concentrations of saccharides such as sucrose, trehalose, lactose etc. and also by polyhydric alcohols such as sorbitol, mannitol, polyethylene glycol etc. due to preferential exclusion from the protein surface, thereby providing stabilisation11. It must be noted that while sucrose is excluded due to an increase in surface tension of water12 and glycerol is excluded by solvophobic effects, polyethylene glycols are excluded because of steric interference11. Similarly, protein solubility can often be increased by adding modest concentrations of salt, a phenomenon known as ‘salting in’. Salts are also used as tonicity modifier; however, in some cases it may negatively impact conformational stability13. Thus, the choice of counterions (with reference to Hofmeister series) and their concentration can be used to alter the stability profile of the biologic. This is considered critical during pre-formulation screening. The same pH condition, for example, can be obtained using different buffer species at various compositions (i.e. at various ionic strengths) and thus can alter the stability of the protein. Ligand binding can also favour the native state of proteins, as seen for example with the binding of Zn2+ to human growth hormone14.
Surface-active agents (e.g. Surfactants) are often added to inhibit adsorption, denaturation and/or aggregation of proteins at interfaces (both air-water and solid-water) and may also impact stability through differential binding to native or denatured states of the protein. Agitation and/or freeze-thaw stress studies are often used to illustrate surface denaturation phenomena and addition of low concentrations of surfactants such as Polysorbate 20, Polysorbate 80, Pluronic F68 or others may be effective to minimise both soluble and insoluble aggregates15. Leachates such as metal ions, barium from glass vials, vulcanising agents from stoppers, tungsten oxide from prefilled syringes and exposure to silicone oil are additional concerns due to primary contact surfaces that need to be addressed during excipient selection. EDTA, for example, can be used to address metal leachates from stoppers16. Thus a deep understanding of the predominant degradation mechanisms of a protein and the use of ligands and co-solutes to enhance its conformational stability can help narrow down the choice of excipients for successful formulation development.
One-third of biological products are available as multidose and these usually require addition of antimicrobial preservatives. The preservatives can present additional stability challenges for biologic products by inducing protein aggregation17. Commonly screened preservatives include m-cresol, benzyl alcohol or phenol, phenoxyethanol and chlorobutanol. Screening of various preservatives either alone or in combination is recommended prior to selection18. Instability of protein in aqueous solution condition for a multidose formulation may force lyophilisation without the preservative and reconstitution with BWFI (Bacteriostatic Water for Injection).
Lyophilised formulation development
Aligning the desired stability, storage and shipping requirements with the target product profile makes lyophilisation a suitable alternative over liquid formulation, especially for highly thermolabile products and live virus vaccine products. The lyophilisation process consists of freezing followed by primary and secondary drying under vacuum. The drying process itself presents an additional set of challenges, as has recently been described19. During freezing, for example, denaturation can occur either in the freeze-concentrate state20, at frozen surface interfaces21 and/or through cold-denaturation22. Thus, the formulation scientist must take into account the impact of local concentrations of salts and buffer and increase in trapped oxygen concentration during the freeze-concentrate state23. Similarly, shifts in pH due to buffer crystallisation must be considered during the formulation design space for lyophilised products24. For example, it is preferable to achieve buffering at physiological pH conditions with low concentration (≤ 10 mM) of potassium phosphate over sodium phosphate buffer. This is attributed to pH shift as a function of increasing buffer concentration and a large pH shift for sodium phosphate during freezing. Citrate, Tris and Histidine are good choices for buffer, if in the appropriate pH range, due to minimal pH shifts during freezing. Similarly, antioxidants (e.g. ascorbates) and scavengers (e.g. thiourea) may be added to limit oxidation25.
Besides cryoprotectants, additional stabilisers known as lyoprotectants might be required. For example, liquid pre-formulation screening might suggest sorbitol as a good stabiliser; however, sorbitol is not preferred in dried formulations due to its low glass transition temperature. It must be noted that high glass transition temperatures to minimise molecular mobility while retaining the native state in the dried state by serving as a good ‘water substitute’ (e.g. sucrose, trehalose) are the two key attributes of lyoprotectants. Additionally, use of reducing sugars (e.g. lactose) must be clearly weighed in the risk-assessment strategy. However, the possibility of phase separation (especially for a multi-stabiliser system), may restrict certain combinations of excipients (e.g. PEG-Dextran)26. Similarly, potential for acid hydrolysis at low pH27 may render trehalose a superior alternative to sucrose. Ease of sourcing sucrose from multiple vendors and low cost compared to trehalose may make sucrose the excipient of choice. Such considerations with a clear line of sight to manufacturing along with the appropriate stability data must be used for excipient selection.
Bulking agents are also added to lyophilised product to prevent product ‘blowout’ for low concentration product (≤ 1% solid), increase the product collapse temperature and/or to improve product elegance. Excipients may serve as amorphous bulking agents (e.g. sucrose, trehalose, lactose, raffinose, dextran, hydroxyethyl starch (HES)) or crystalline bulking agents (e.g. glycine, mannitol). Excipients with high glass transition temperatures (Tg’) for amorphous excipients (e.g. HES) or high eutectic temperatures (Teu) for crystalline excipients (e.g. glycine and mannitol) allow for faster primary drying. However, presence of hydrate form of mannitol and possibility of glass breakage during manufacturing (either due to high fill volume, incorrect freezing protocol and/or high concentration) might limit the selection of mannitol during excipient screening.
Adjuvants in vaccine formulation
Vaccine formulations often include an additional component for immune-enhancement called adjuvants. Adjuvants are more commonly used for subunit, protein and peptide-based antigens and help enhance the intrinsic immunogenicity of the antigens28. Aluminium-based adjuvants such as aluminium hydroxide (Alhydrogel) and aluminium phosphate (AdjuPhos) are the most common class of adjuvants for human use due to their historic safety and efficacy profiles. Historically the binding of antigens to aluminium based adjuvants was assumed to be important, although a number of recent studies suggest that antigen–adjuvant binding may not always be necessary29. It is important to understand the antigen-adjuvant interactions and their impact on stability and immunogenicity before finalising the type and amount of adjuvant to be used. The inclusion of aluminium adjuvants may warrant inclusion of excipients that stabilise antigen-adjuvant complexes and prevent physical and chemical degradation of the antigen on the surface of the adjuvants30,31.
Conclusions
This review provides a general overview and approach to excipient screening for biologic and vaccine formulations. The excipients should be selected based on a comprehensive understanding of their impact on product during multiple events that may encompass drug substance processing, freeze-thaw, time-in-solution, filtration, mixing, material compatibility, formulation/fill process, lyophilisation process, long-term stability, photosensitivity, time-out of refrigeration (TOR), in-use stability (post-reconstitution stability) and shipping. Excipients must be suitable for the intended market and compendial (USP/NF, EP, JP) grades must be used when possible. Biologics and vaccines are mostly formulated for parenteral delivery and require endotoxin-free excipients. The cost and ease of sourcing pharmaceutical grade excipients should also be considered before finalising the excipients.
References
- Kamerzell, TJ. et al. Protein-excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv. Drug Del. Reviews, 63:1118-1159 (2011)
- Kwissa M, Kasturi SP, Pulendran B. The science of adjuvants. Expert Rev. Vaccines, 6(5):673-84 (2007)
- EMA Guideline on excipients in the dossier for application for marketing authorisation of a medicinal product, (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003382.pdf), Jan (2008)
- FDA guidance: Guidance for Industry Nonclinical Studies for the Safety Evaluation of Pharmaceutical Excipients, (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079250.pdf), May (2005)
- Hardeep, S.; Feng, H.; Bhambhani, A.; Pipkins, J.; Zimmerer, R.; Joshi, S.; Middaugh, C. The Effects of Substituted Cyclodextrins on the Colloidal and Conformational Stability of Selected Proteins. J. Pharm. Sci. 99(6), 2800-2818 (2010)
- Bhambhani, A.; Kissmann, J. M.; Joshi, S. B.; Volkin, D. V.; Kashi, R. S.; Middaugh, C. R. Formulation Design and High Throughput Excipient Selection Based on Structural Integrity and Conformational Stability of Dilute and Highly-Concentrated IgG1 Monoclonal Antibody Solutions. J. Pharm. Sci. 101(3), 1120-1035 (2011)
- Chen D. and Kristensen, D. Opportunities and challenges of developing thermostable vaccines. Expert Rev. Vaccines, 8(5): 547-557 (2009)
- ICH (Int. Conf. Harmonization) Pharmaceutical Development, Q8, Aug (2009)
- Akers, M. J.; Vasudevan, V.; Stickelmeyer, M. Formulation development of protein dosage forms, in Development and Manufacture of Protein Pharmaceuticals, Vol. 14 of Pharmaceutical Biotechnology Series, Nail, S. L.; Akers, M. L., eds. Plenum, New York, pp. 47-127 (2002)
- Bhambhani, A.; Thakkar, S.; Joshi, S. B.; Middaugh, C. R. A formulation method to improve the physical stability of macromolecular-based drug products. Meyer, B. K. (Eds.) Therapeutic protein drug products: Practical approaches to formulation in the laboratory, manufacturing, and the clinic (2012)
- Timasheff, S. N. Preferential interactions of water and cosolvents with proteins, Chapter 11 in: Protein Solvent Interactions, Robert, B.; Gregory, ed. Marcel-Dekker, New York, pp. 445-482 (1995)
- Lee, J. C. and Timasheff, S. N. The stabilization of proteins by sucrose. Biol. Chem. 256, 7193–7201 (1981)
- Moore, J.M.; Patapoff, T. W.; Cromwell, M. E. Kinetics and thermodynamics of dimer formation and dissociation for a recombinant humanized monoclonal antibody to vascular endothelial growth factor. Biochemistry, 38(42), 13960-13967 (1999)
- Cunningham, B. C., Mulkerrin, M. G.; Wells, J. A. Dimerization of human growth-hormone by zinc. Science 253(5019), 545-548 (1991)
- Levine, H.L.; Ransohoff, T.C.; Kawahata, R.T.; McGregor, W. C. The use of surface tension measurements in the design of antibody-based product formulations. J. Parenter. Sci. Technol. 45(3), 160-165 (1991)
- Marcovic, I. Challenges associated with extractable and/or leachable substances in therapeutic biologic protein products. Am. Pharm. Rev. (Sept./Oct.), 20-27 (2006)
- Maa YF, Hsu CC. Aggregation of recombinant human growth hormone induced by phenolic compounds. Int J Pharm140, 155–16 (1996)
- Hutchings RL, M Singh S, Cabello-Villegas J, Mallela KM. Effect of antimicrobial preservatives on partial protein unfolding and aggregation. J Pharm Sci. 102(2), 365-376 (2013)
- Bhambhani, A.; Blue, J. T. Lyophilization Strategies for Development of a High-Concentration Monoclonal Antibody Formulation: Benefits and Pitfalls, Am. Pharm. Rev. 31-38 (2010)
- Hatley, R. H. M.; Mant, A. Determination of the unfrozen water content of maximally freeze concentrated carbohydrate solution Int. J. Biol. Macromol. 15, 227-232 (1993).
- Strambini, G. B.; Gabellieri, E. Proteins in frozen solutions: Evidence in ice-induced partial unfolding. Biophys. J. 70(2), 971-976 (1996)
- Privalov, P. L. Cold denaturation of proteins. Crit. Rev. Biochem. Mol. Biol. 25, 281-305 (1990)
- Murase, N.; Franks, F. Salt precipitation during the freeze-concentration of phosphate buffer solutions. Biophys. Chem. 34, 293-300 (1989)
- van den Berg, L.; Rose, D. Effect of freezing on the pH and composition of sodium and potassium phosphate solutions: The reciprocal system: KH2PO4-Na2HPO4-H2O, Arch. Biochem. Biophys. 81, 319-329 (1959)
- Chang, B.S. and Hershenson, S. Practical approaches to protein formulation development. in “Rationale Design of stable protein formulations-theory and practice” (J.F. Carpenter and M.C. Manning eds.) Kluwer Academic/Plenum publishers, New York, pp. 1-25 (2002)
- Gomez, G.; Pikal, M. J.; Rodriguez-Horned, N. Effect of initial buffer composition on pH changes during far-from equilibrium freezing of sodium phosphate buffer solutions. Pharm Res. 18, 90-97 (2001)
- Banks DD, et al. The Effect of Sucrose Hydrolysis on the Stability of Protein Therapeutics During Accelerated Formulation Studies. Pharm. Sci. 98(12), 4501–4510 (2009)
- Guy, The perfect mix: recent progress in adjuvant research, Nat. Rev. Microbiol. 5, 505–517 (2007)
- Z. Romero Mendez, Y. Shi, H. HogenEsch, S.L. Hem, Potentiation of the immune response to non-adsorbed antigens by aluminum-containing adjuvants, Vaccine 25, 825–833 (2007)
- J. Peek, T.T. Martin, C. Elk Nation, S.A. Pegram, C.R. Middaugh, Effects of stabilizers on the destabilization of proteins upon adsorption to aluminum salt adjuvants, J. Pharm. Sci. 96, 547–557 (2007)
- S. Jones, L.J. Peek, J. Power, A. Markham, B. Yazzie, C.R. Middaugh, Effects of adsorption to aluminum salt adjuvants on the structure and stability of model protein antigens, J. Biol. Chem. 280, 13406–13414 (2005)
Biographies
Babu Medi, PhD is an Associate Principal Scientist at Merck & Co, Inc., (WestPoint, PA). He received his Bachelor’s degree in Pharmaceutical Sciences (1999) from Andhra University, India and PhD in Pharmaceutical Sciences (2005) from North Dakota State University (Fargo, ND). His current research involves vaccine drug product development for novel vaccine candidates including liquid, lyophilised and adjuvanted formulations. Prior to joining Merck in 2009, Babu worked on novel vaccine delivery technologies aimed at developing needle-free and room temperature stable vaccine formulations at DelSite Biotechnologies (2005 – 2009).
Ramesh Chintala, PhD received his PhD (1997) in Chemistry from University of Madras, India. He is currently an Associate Principal Scientist supporting vaccine pre-formulation and drug product development within Vaccine Bioprocess R&D at Merck & Co., Inc, (West Point). He previously held positions at Enzon Pharmaceuticals working on Pegylation of proteins, downstream processing and analytical characterisation. Prior to joining the pharmaceutical industry, Ramesh worked as a postdoctoral fellow at Kyoto Pharmaceutical University (Kyoto, Japan) and later moved to Rutgers University (Piscataway, NJ) for his second post-doctoral fellowship. His areas of expertise include biologics and vaccine / adjuvant formulation development that employs the application of material sparing methods and surface characterisation of pharmaceutical materials.
Akhilesh Bhambhani, PhD is an Associate Principal Scientist at the Merck & Co., Inc., (West Point, PA). He earned his BS in Chemical Engineering in 2001 from Ujjain Engineering College, India and his PhD in Biological Chemistry in 2006 from University of Connecticut (Storrs, CT). Prior to joining Merck in 2009, Akhilesh worked as a postdoctoral researcher at The University of Kansas (Lawrence, KS) and later as a Development Scientist I at Amylin Pharmaceuticals (San Diego, CA) developing various liquid and lyophilised products for biologics including high concentration antibody formulation and formulations of peptides, proteins, bacteria, and vaccines. In his current role, he is leading a team focused on evaluation and development of novel drying and manufacturing technologies for high volume / low cost manufacturing of live-virus vaccines.




useful