

Molecular drug targets in autophagy
Posted: 19 April 2011 |
Autophagy is a cellular stress response to diverse stimuli such as starvation, infection and DNA damage. Autophagy plays important roles in the progression of various diseases including cancer, neurodegenerative diseases and Crohn’s disease. Despite recent advances in our understanding of the autophagy machinery, surprisingly little effort has been undertaken towards utilising this knowledge in drug discovery processes. Several phenotypic screens have been undertaken to identify drug candidates that modulate this process. Current highthroughput screening approaches assay the formation of the autophagosome and very little effort is made towards the identification of compounds that inhibit specific autophagy components. Here, I give an overview about potential molecular drug targets in the autophagy pathway and review the current status of targeted drug discovery towards identifying autophagy gene-specific drugs.
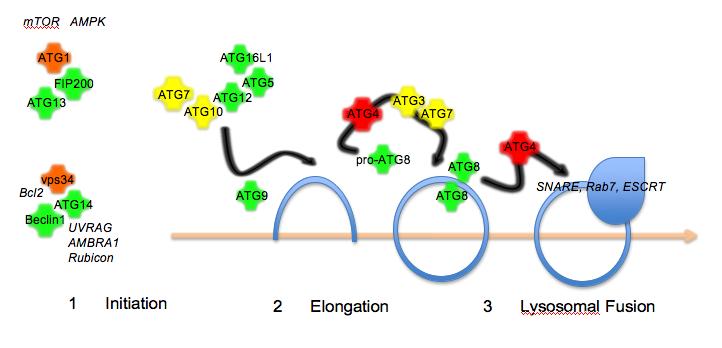
Figure 1The autophagy pathway. Autophagy proceeds in three steps: initiation by protein kinase complexes
Autophagy is a cellular stress response to diverse stimuli such as starvation, infection and DNA damage. Autophagy plays important roles in the progression of various diseases including cancer, neurodegenerative diseases and Crohn’s disease. Despite recent advances in our understanding of the autophagy machinery, surprisingly little effort has been undertaken towards utilising this knowledge in drug discovery processes. Several phenotypic screens have been undertaken to identify drug candidates that modulate this process. Current highthroughput screening approaches assay the formation of the autophagosome and very little effort is made towards the identification of compounds that inhibit specific autophagy components. Here, I give an overview about potential molecular drug targets in the autophagy pathway and review the current status of targeted drug discovery towards identifying autophagy gene-specific drugs.
Macroautophagy (hereafter termed autophagy) is a cellular stress response that results in activation of a lysosomal degradation pathway. Autophagy has implications in diverse contexts important for human health, such as bacterial or viral infection1,2, neurodegeneration3-5 and cancer6-9. Targeting the autophagy pathway may be a therapeutic opportunity to treat disease states in these conditions10.
In recent years, the components of the autophagy machinery have been identified in yeast, flies, worms and mammalian cells. In yeast, this currently includes over 30 members that fall into three broad categories: (1) initiation of biogenesis of the membrane (2) elongation of the isolation membrane (3) final closure of the autophagosome and fusion with the lysosome (Figure 1). Most of the human homologues have been identified, though there are still some not yet characterised. In addition, metazoan-specific genes involved in autophagy, some of which have mammalian counterparts, have been identified11. Autophagy initiation requires two signalling complexes, both of which include a protein kinase, ATG1 (in complex with ATG13, ATG101 and FIP200) and vps34 (PI3K class III; in complex with Beclin1/ATG6, ATG14 and p150). Elongation requires the transmembrane protein ATG9 and a complex composed of ATG5, ATG12 and ATG16L1 that is formed by the E1 ubiquitin conjugating enzyme-like ATG7 and the E2 ubiquitin conjugating enzyme-like ATG10. Another E2 ubiquitin conjugating enzyme-like ligase, ATG3, in conjunction with ATG7 is involved in processing of the autophasomal membrane-associated marker protein ATG8. In addition, ATG8 requires proteolytic cleavage by members of the ATG4 family. Several human ATG8 isoforms have been identified (hMAP1LC3, GATE16, GABARAP) and GFP- or RFP-tagged versions have been used as a marker for autophagosome formation. Fusion of autophagosomes with lysosomes requires the ESCRT comples12, dynein13, SNAREs14, Rab715 and class C vps proteins16,17. In addition, lysosomal integrity and acidification is a requirement for fusion with the autophagosome.

Figure 1The autophagy pathway. Autophagy proceeds in three steps: initiation by protein kinase complexes
Autophagy in disease
Genomic data have identified several autophagy gene defects in the onset of diseases including ATG16L1 in Crohn’s disease18,19, ATG7 in Huntington’s disease20, and Beclin1 in tumour formation21,22.
From studies using knockout mice, it has become evident that autophagy is involved in embryonic development, neonatal survival, erythrocyte and lymphocyte differentiation and prevention of neuro-degeneration (reviewed in Mizushima and Levine23). In addition, it can be expected that autophagy controls long-term homeostasis of cells in general, which has not been properly addressed in experimental models so far. It should be noted that genetic loss of essential autophagy genes does not inevitably lead to a complete absence of autophagosomes. Table 1 gives an overview of autophagosomal defects in cell lines upon loss of autophagy genes. Loss of factors involved in the initiation of membrane biogenesis such as ATG9 and FIP200 lead to very low or undetectable levels of autophagosomes24,25 while factors involved in the conjugation of LC3 such as ATG3 and ATG4B generally show defects in autophagosome maturation26,27. It should be noted that alternative pathways (i.e. ATG5- and ATG7-independent) for autophagosome formation have been identified28 that may lead to functional autophagosomes. Not all of the knockout cell lines have been examined with standardised assays. For example, fluorescence microscopy of GFP-LC3 puncta has limitations in the resolution of individual autophagosomes and should be supported by high-resolution electron microscopy in order to identify changes in autophagosome maturation. In terms of the therapeutic benefit, it needs to be formally addressed whether a block in initiation, maturation or lysosomal fusion of the autophagosome is beneficial and this may well vary between different disease models or tissues.
Table 1: Autophagosome formation in autophagy gene (ATG) defective cell lines
| Gene | Autophagosomes | Mutant | Reference |
| ULK1 | Reduced | Knockout mouse | (43, 56) |
| ATG3 | Incomplete closure | Knockout mouse | (27) |
| ATG5 | Alternative autophagosomes | Knockout mouse | (28) |
| ATG7 | Alternative autophagosomes | Knockout mouse | (28) |
| ATG9 | Smaller autophagosomes | Knockout mouse | (24) |
| ATG16L1 | Incomplete closure | Knockout mouse | (57) |
| ATG4B | Incomplete closure | Dominant negative | (26) |
| ATG4B | Normal | Knockout mouse | (48) |
| ATG4C | Normal | Knockout mouse | (46) |
| FIP200 | Not detectable | Knockout mouse | (25) |
| VPS34 | Alternative autophagosomes | Knockout mouse | (41) |
| ATG14L1 | Reduced number | Knockout mouse | (58) |
Status of drug discovery in autophagy
Many efforts aim to identify novel small molecule therapeutics that target components of the autophagy pathway29,30. Most commonly, activators of autophagy are considered of value for neurodegenerative diseases, whereas inhibitors of autophagic flux such as chloroquine are considered effective in cancer therapy. Several reports have shown high combinatorial effects of chloroquine with first-line therapeutics in chemotherapy-resistant cell lines31-34. These studies support the notion that targeting autophagy provides a therapeutic benefit in models of chemotherapy resistance. Some clinical trials are underway that address the efficiency of chloroquine or hydroxychloroquine treatment in combination with other cytostatic drugs (Table 2). Based on the role of autophagy as a tumour suppressive mechanism21,22 in early stages of tumour formation, there may be a benefit in identifying activators of autophagy as anticancer agents as well.
Table 2: Clinical trials based on inhibition of autophagy
| Compound | Indication | Phase | ClinicalTrials.gov identifier |
| Chloroquine | Small Lung Cell Cancer | I | NCT00969306 |
| Hydroxychloroquine (combination treatment) | Non-Small Lung Cell Cancer | II | NCT00933803 |
| Hydroxychloroquine | Melanoma (skin) | I | NCT00962845 |
| Hydroxychloroquine + Ixabepilone | Breast Cancer | II | NCT00765765 |
| Hydroxychloroquine (combination treatment) | Rectal Cancer, Colon Cancer, Metastasis, Adenocarcinoma | II | NCT01206530 |
| Hydroxychloroquine | Renal Cell Carcinoma | I | NCT01144169 |
| Chloroquine + Tamoxifen | Carcinoma, intraductal | II | NCT01023477 |
| Hydroxychloroquine | Unspecified Adult Solid Tumor | I | NCT00909831 |
| Hydroxychloroquine + Docetaxel | Prostate Cancer | II | NCT00786682 |
| Hydroxychloroqine (combination treatment) | Colorectal Cancer | II | NCT01006369 |
| Hydroxychloroquine (combination treatment) | Advanced Cancer | I | NCT01266057 |
| Hydroxychloroquine + Vorinostat | Advanced Solid Tumor | I | NCT01023737 |
| Hydroxychloroquine (combination treatment) | Lung Cancer | II | NCT00728845 |
| Hydroxychloroquine + Bortezomib | Multiple Myeloma and Plasma Cell Neoplasm | II | NCT00568880 |
| Hydroxychloroquine + Gemcitabine | Pancreatic Cancer | II | NCT01128296 |
Molecular targets for pharmaceutical intervention in the autophagy pathway fall into two categories: they can be components of the autophagy machinery itself or alternatively they can be upstream regulators such as signal transduction cascades that modulate the activity of the autophagy machinery.
Chemical screening approaches have identified a number of drugs that elicit increases in autophagic responses such as autophagosome formation or aggregated protein clearance35,36 and most of these compounds target upstream signalling molecules. Overall, targeting components of the autophagy machinery itself is a highly neglected area of drug discovery research.
Molecular drug targets
The most obvious candidates for drug targeting within the autophagy machinery are enzymes or protein-protein interactions. Multiple enzymes operate in the autophagy pathway and these include kinases (ATG1, vps34), proteases (ATG4A-D) and ubiquitin conjugating enzymelike ligases (ATG3, 7, 10).
Kinases
The two major kinases that operate in autophagy are vps34 (PI3K class III) and ATG1 (human ULK1/2). An ongoing effort has been to identify specific inhibitors of vps34 (PI3K class III). So far, this has not been successful, probably due to the fact that the adenine-binding pocket is constricted37. Current inhibitors of vps34 such as 3-MA lack the specificity to be of clinical use and have poor pharmacological properties. Recent crystallography studies of vps34 have identified promising new approaches to identify inhibitors37 that will need to be explored in drug discovery research further. Interestingly, the Beclin1-vps34 complex is differentially regulated by Rubicon and AMBRA1 or UVRAG resulting in either inhibition or activation of autophagy38-40. Disruption of these complexes may provide an opportunity for drug targeting. Of note is a vps34-independent pathway for autophagosome formation that has been reported in knockout animals raising concerns with regards to molecular redundancies41. As stated above, it needs to be explored whether modulation of autophagosome maturation or a complete absence of autophagosomes provides the highest therapeutic benefit.
ATG1 is required for multiple steps in autophagosome formation and is under control of signals emanating from mTOR42 and AMPK43. Interestingly, mTOR inhibits ULK1 while AMPK promotes autophagy by phosphorylation of ULK143. Based on promising results with PI3K and mTOR inhibitors as anti-cancer agents, targeting ATG1 may provide a more autophagy-specific therapeutic opportunity within this signalling cascade. A crystal structure for ATG1/ULK1 has not yet been reported, and it is not clear at the moment whether the active site is accessible to small molecule compounds.
Proteases
ATG4 seems a likely candidate for pharmacological intervention as it is a key enzyme involved in two essential steps involved in LC3 processing. The availability of highthroughput screening methods both in vitro44 and in cell-based systems45 makes this family a very attractive therapeutic target. Loss of ATG4C results in enhanced sensitivity for fibrosarcoma46, further supporting the notion that members of this family are relevant in disease settings. The crystal structure of ATG4B shows that the catalytic site is buried in a pocket and unlikely to be accessible for small molecule compounds47. An N-terminal activation loop undergoes conformational change upon binding to LC3, thus offering a therapeutic opportunity to target this specific interaction. The mild phenotype of ATG4B knockout mice and the presence of normal autophagosomes in both ATG4B and ATG4C knockout mice46,48 suggests that there are some redundancies in this protein family that need to be taken into account for drug design.
Uqibuitin-like Ligases
Ubiquitin-conjugating enzymes have recently been recognised as promising molecular drug targets49,50. There are three ubiquitin conjugating enzyme-like proteins in autophagy: ATG7 (E1-like enzyme), ATG3 and ATG10 (both E2-like enzymes).
Given the essential role of ATG7 for both conjugation complexes, it is reasonable that interference with its catalytic activity may result in an inhibition of autophagosome formation, although alternative mechanisms for autophagosome formation exist28. The disease relevance is underscored by the fact that a single nucleotide polymorphism in ATG7 is associated with onset of Huntington’s disease20. There are two common mechanisms for small molecule inhibition of E1 ubiquitin-conjugating enzymes: covalent inactivation of the active site or inhibition of conjugation via adduct formation50. In yeast, the ATG7 homolog has been shown to form homodimers via the C-terminal region that is required for its protein conjugating activity towards ATG12 and ATG851. Thus, blocking dimerization may be another potential therapeutic strategy. In the absence of a crystal structure, it is not clear how accessible these sites are for small molecule compounds.
E2 conjugating enzymes can be targeted by covalent modification of the active site or by disruption of recognition of the conjugation target50. A crystal structure for ATG3 shows that the catalytic cysteine residue is exposed and accessible to small molecule inhibitors52. ATG3 knockout mice do not survive the neonatal starvation period and show incomplete closure of autophagosomes in embryonic fibroblasts27 thus supporting the notion that this is an essential component of the autophagy machinery and a likely candidate for drug targeting. Interestingly, disruption of ATG12 binding to ATG3 does not affect general autophagy, but is rather required for mitochondrial homeostasis53. The ubiquitin-like conjugating enzyme ATG10 is required for binding of ATG12 to ATG5, an essential step in the formation of autophagosomes. Interestingly, a mutation of the catalytic cysteine of ATG10 results in a block of ATG5-12 conjugation54, thus indicating that targeting this site may be beneficial. A preliminary crystal structure has been reported55, but not fully analysed. Thus, it is not clear at the moment whether the catalytic site is accessible to small molecule compounds.
Conclusions
Current clinical trials using inhibitors of autophagosome/lysosome fusion show very promising results in combinatorial cancer treatment. Other potential molecular targets within the PI3K-mTOR pathway show great promise as well, although it is not clear whether these effects are linked to de-regulation of autophagy or a direct effect of inhibiting the respective signalling pathway. A potential new route for drug discovery could be the identification of small molecule compounds that directly target components of the autophagy machinery. The most promising candidates are ATG1 (ULK1/2), ATG3, ATG4B, ATG7, ATG10 and vps34 (PI3K III). It is possible that treatment of late-stage and chemoresistant tumours may benefit from a block in autophagosomal maturation, while in neurodegenerative diseases, a general activation of autophagy may be beneficial. A lot more research needs to be aimed at drug discovery in autophagy to translate the rapidly growing basic research findings to a therapeutic tractable model system.
References
1. M. I. Colombo (2005) Cell Death Differ 12 Suppl 2, 1481
2. W. T. Jackson et al., (2005) PLoS Biol 3, e156
3. D. J. Klionsky, (2006) Nature 441, 819
4. U. B. Pandey et al., (2007) Nature 447, 859
5. M. Komatsu et al., (2007) Proc Natl Acad Sci U S A 104, 14489
6. R. Mathew, V. Karantza-Wadsworth, E. White, (2007) Nat Rev Cancer 7, 961
7. K. Degenhardt et al., (2006) Cancer Cell 10, 51
8. D. C. Rubinsztein, J. E. Gestwicki, L. O. Murphy, D. J. Klionsky, (2007) Nat Rev Drug Discov 6, 304
9. Y. Takahashi et al., (2007) Nat Cell Biol 9, 1142
10. R. K. Amaravadi, C. B. Thompson, (2007) Clin Cancer Res 13, 7271
11. Y. Tian et al., (2010) Cell 141, 1042
12. D. Metcalf, A. M. Isaacs, (2010) Biochem Soc Trans 38, 1469
13. L. Jahreiss, F. M. Menzies, D. C. Rubinsztein, (2008) Traffic 9, 574
14. M. Renna et al., (2011) Journal of cell science 124, 469
15. S. Pankiv et al., (2010) J Cell Biol 188, 253
16. N. Furuta, T. Yoshimori, A. Amano, (2010) Autophagy 6, 417
17. Longatti, S. A. Tooze, (2009) Cell Death Differ 16, 956
18. J. D. Rioux et al., (2007) Nat Genet 39, 596
19. J. Hampe et al., (2007) Nature genetics 39, 207
20. S. Metzger et al., (2010) Hum Genet 128, 453
21. X. Qu et al., (2003) J Clin Invest 112, 1809
22. Z. Yue, S. Jin, C. Yang, A. J. Levine, N. Heintz, (2003) Proc Natl Acad Sci U S A 100, 15077
23. N. Mizushima, B. Levine, (2010) Nat Cell Biol 12, 823
24. T. Saitoh et al., (2009) Proc Natl Acad Sci U S A 106, 20842
25. T. Hara et al., (2008) J Cell Biol 181, 497 26. N. Fujita et al., (2008) Mol Biol Cell 19, 4651
27. Y. S. Sou et al., (2008) Mol Biol Cell 19, 4762
28. Y. Nishida et al., (2009) Nature 461, 654
29. S. Sarkar et al., (2007) Nat Chem Biol 3, 331
30. L. Zhang et al., (2007) Proc Natl Acad Sci U S A 104, 19023
31. M. Degtyarev et al., (2008) J Cell Biol 183, 101
32. S. Chen et al., (2010) Biochim Biophys Acta 1806, 220
33. K. Sasaki et al., (2010) BMC Cancer 10, 370
34. C. Bellodi et al., (2009) J Clin Invest 119, 1109
35. B. Ravikumar et al., (2010) Physiol Rev 90, 1383
36. S. Sarkar et al., (2007) Nat Chem Biol 3, 331
37. S. Miller et al., (2010) Science 327, 1638
38. E. Itakura, C. Kishi, K. Inoue, N. Mizushima, (2008) Mol Biol Cell 19, 5360
39. G. M. Fimia et al., (2007) Nature 447, 1121
40. K. Matsunaga et al., (2009) Nat Cell Biol 11, 385
41. X. Zhou et al., (2010) Proc Natl Acad Sci U S A 107, 9424
42. D. F. Egan et al., (2011) Science 331, 456
43. J. Kim, M. Kundu, B. Viollet, K.-L. Guan, (2011) Nat Cell Biol 13, 132
44. C. W. Shu et al., (2011) J Biomol Screen 4
5. R. Ketteler, Z. Sun, K. F. Kovacs, W.-W. He, B. Seed, (2008) Genome Biol 9, R64
46. G. Marino et al., (2007) J Biol Chem 282, 18573
47. K. Satoo et al., (2009) Embo J 28, 1341
48. G. Marino et al., (2010) J Clin Invest 120, 2331
49. S. R. Ande, J. Chen, S. Maddika, (2009) Eur J Pharmacol 625, 199 5
0. L. Bedford, J. Lowe, L. R. Dick, R. J. Mayer, J. E. Brownell, (2011) Nat Rev Drug Discov 10, 29
51. M. Komatsu et al., (2001) J Biol Chem 276, 9846
52. Y. Yamada et al., (2007) J Biol Chem 282, 8036
53. L. Radoshevich et al., (2010) Cell 142, 590
54. T. Nemoto et al., (2003) J Biol Chem 278, 39517
55. M. Yamaguti, N. N. Suzuki, Y. Fujioka, Y. Ohsumi, F. Inagaki, (2007) Acta Crystallogr Sect F Struct Biol Cryst Commun 63, 443
56. M. Kundu et al., (2008) Blood 112, 1493
57. T. Saitoh et al., (2008) Nature 456, 264
58. K. Matsunaga et al., (2010) J Cell Biol 190, 511
About the Author
Robin Ketteler studied bio – chemistry at the Free University Berlin and did his PhD at the Max-Planck-Institute for Immunobiology. As postdoctoral fellow at MGH in Boston, he used highthroughput screening approaches to study cellular signaling pathways. Since 2009, he manages the Translational Research Resource Centre at MRC LMCB in London.
