Options for quantitative analysis by real-time PCR
Posted: 11 November 2005 | | No comments yet
The expansion of microarray-based gene expression studies has led to an increase in demand for gene-specific PCR-based methods for independent validation of results. Although a number of technologies are available to meet this requirement the most popular is currently real-time PCR.
Real-time PCR was first described in the early 1990s1 and uses fluorescent dyes to monitor the accumulation of the PCR product in ‘real-time’. The technique is currently considered the ‘gold standard’ for quantitative PCR methodology – mainly due to its sensitivity (single figure magnitudes of transcript copy number can be detected), specificity, cost-effectiveness, simplicity (no further post-processing is required), relatively high throughput and high dynamic range for quantifying large degrees of differential regulation (> 107 fold)2. It is these features of the technique that often lead to preferential implementation over other quantitative assays such as Northern blot, in situ hybridisation and RNase protection assays.
Most commonly, real-time PCR is employed as a tool to determine relative gene expression, although less frequently used applications of absolute expression quantitation and genotyping have been demonstrated. These have been shown to have a wide variety of applications including the validation of alternative quantitation technologies (e.g. microarray data)3, molecular diagnostics (e.g. determination of viral and bacterial loads)4, alleleotyping5, forensics and identifying DNA copy number alterations6.
Within this review various technological aspects of real-time PCR will be considered, including detection chemistries and thermocycler options.
Detection chemistries
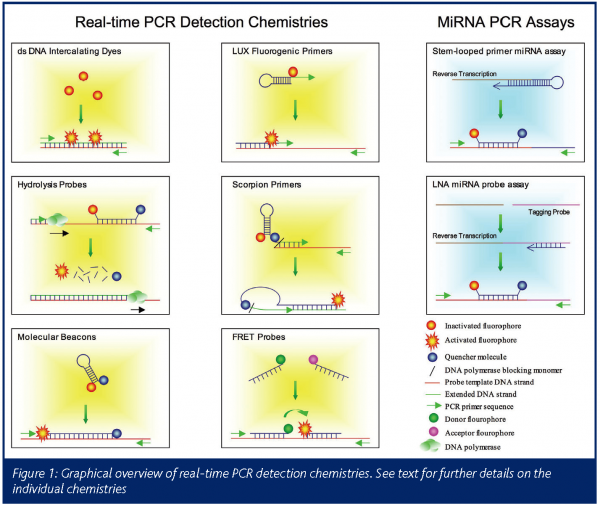
Whilst there are a number of methods for determining both absolute and relative cDNA or DNA starting amounts, all rely on the determination of the threshold cycle, Ct (for a review of analytical procedures see www.gene-quantification.info). This is described as the PCR cycle where the accumulation of the PCR product is first detectable above background levels and is inversely proportional to the starting amount of template. Methods for detecting the PCR product accumulation can be broadly defined as fluorescent dyes that intercalate into double stranded DNA (dsDNA) or transcript specific fluorescent-tagged hybridisation probes that anneal to the PCR product in a sequence-specific manner (Figure 1).
Intercalating dyes
Intercalating dyes such as SYBR Green I (Molecular Probes) emit up to 1000-fold greater fluorescence when bound to the minor groove of dsDNA than single stranded DNA. They are the most flexible of the detection chemistries because they act in an assay-independent fashion. Thus the only specific requirements for the assay are standard PCR primers and a real-time PCR thermocycler to measure fluorescence accumulation. Whilst this results in a relatively inexpensive assay, the main disadvantages arise because the dye will incorporate into any dsDNA that accumulates as part of the PCR process. Consequently, different assays cannot be multiplexed and are sensitive to the accumulation of non-specific products and primer-dimers. Each assay must therefore be individually optimised to ensure that only the product of interest is amplified. Non-specific products can be detected by performing a melt-curve analysis at the end of the PCR stage. The accumulated products are subjected to an increasing temperature gradient over time, such that the products gradually become single stranded in a sequence specific manner. The rate of associated decrease in fluorescence can be detected and presence of non-specific products with different sequence and therefore different melting characteristics can be determined.
Hybridisation probes
In contrast to the intercalating dyes, hybridisation probes are fluorescently labelled oligonucleotides designed to bind specifically to the transcript of interest. These can either be incorporated into the design of a PCR primer (exemplified by the Scorpion and LUX technologies) or act as one or two additional sequence specific oligonucleotides that bind to the PCR amplified sequence (exemplified by TaqMan hydrolysis probes, Applied Biosystems). Although the attachment of fluorophores to the probes results in a considerable increase in assay cost, the use of gene specific probes has a number of advantages over intercalating dyes: (i) different coloured fluorophores can be used to detect each transcript, thus allowing several transcripts to be amplified and detected in the same multiplexed reaction; (ii) the probes should only bind to the PCR product of interest and therefore offer greater specificity than intercalating dyes; (iii) sequence specificity enables the detection of allelic differences.
Hydrolysis probes
Also known as the 5’ nuclease assay, hydrolysis probes – as exemplified by the TaqMan chemistry – consist of a sequence specific probe with a fluorophore attached to the 5’ end and a quencher attached to the 3’ end. When the probe is not hybridised to the target, the quencher molecule reduces the fluorescent emission by fluorescence resonance energy transfer (FRET). Upon target binding, the 5’ nuclease activity of the DNA polymerase cause the degradation of the probe during the PCR extension step and this results in increased fluorescence due to a separation of the fluorophore and quencher molecules.
Molecular beacons
In similarity to the TaqMan chemistry, molecular beacons are sequence specific probes with a 5’ fluorophore and 3’ quencher molecule. They are designed with inverted repeat sequences on each end, which hybridise together resulting in a hairpin-loop confirmation of the probe. This effectively brings the fluorophore and quencher molecules in close proximity and an associated reduction in fluorescence. As the probe binds to the target sequence, the probe opens out and an increase in fluorescence can be detected due to the separation of the fluorophore and quencher molecules.
FRET probes
Two sequence specific probes are used that are designed to anneal to the target in a head-to-tail fashion. One probe is labelled with a donor fluorophore and the other with an acceptor fluorophore. When the probes are brought to close proximity as a result of target binding, energy transfer from the donor to acceptor fluorophores by FRET causes an increase in fluorescence. Whilst the use of two probes results in an increase in specificity, this also leads to an increase in assay costs.
Sunrise primers
Sunrise primers (Oncor) use similar hairpin loop technology to molecular beacons except that the probe also acts as one of the PCR primers. When the 3’ end of the hairpin loop is used to prime PCR synthesis, the oligonucleotide becomes incorporated into the PCR product, resulting in a separation of the fluorophore and quencher molecules with an associated increase in fluorescence.
Scorpion probes
Scorpion probes (Eurogentec) also combine the detection probe and the upstream PCR primer. The oligonucleotide structure comprises a 5’ fluorophore; a complementary stem loop structure that contains a sequence specific probe sequence within the loop; a quencher dye; a monomer that prevents DNA polymerase extension and 3’ PCR primer sequence. In a standard conformation the oligonucleotide does not fluoresce, due to quenching of the fluorophore by the quencher molecule. Upon binding to the target, the 3’ primer sequence is extended by DNA polymerase and the target specific probe sequence within the stem loop structure binds to the PCR product causing a change in the conformation of the oligonucleotide and an increase in fluorescence.
LUX fluorogenic probes
LUX (Light upon Extension) (Invitrogen) probes are similar to Sunrise primers but do not require a quencher molecule and are therefore less expensive than other hybridisation probe technologies. In the standard conformation, the fluorescence is instead suppressed by the secondary structure of the 3’ end of the oligonucleotide.
Locked Nucleic Acid (LNA) probes
The locked nucleic probes use the same methodology as hydrolysis probes. However, the probes are synthesised with nucleic acid analogues where a methylene bridge connects the 2’-O atom with the 4’-C atom resulting in a ‘locked’ ribose ring. The effect of these locked nucleic acids is to increase the melting temperature (Tm) of the probe and also enable greater differentiation of the Tm of probes with similar sequence. As a result the probes are only 8-9bp in length and, thus, are able to bind to multiple mRNA targets. Using an optimal and bioinformatically designed PCR primer target, the whole genome can be detected using a set of only 90 different probes (Universal ProbeLibrary, Roche). This means that the assays are much less expensive than gene-specific probes with relatively minor differences in specificity.
miRNA probes
Recent evidence is increasingly implicating the role of micro RNAs (miRNA) in many development and disease processes. These are short (20-25bp) – apparently non-coding – RNA that are processed by cleavage of larger (~70bp) precursor molecules. An absence of a poly-A tail and the short nature mean that they are not amenable to conventional real-time PCR detection techniques. The requirement for quantitative profiling assays, however, has led to the development of at least two assay types.
Applied Biosystems have developed a strategy that uses a stem-looped oligonucleotide to prime reverse transcription and the resulting cDNA is PCR amplified and detected using conventional TaqMan probes (Figure 1). The advantage of this strategy is that only mature, cleaved forms of the miRNA is detected and not the precursor molecule. The miRCURY technology (Exiqon) attaches a tagging probe on to the end of the miRNA and this sequence is used to prime cDNA synthesis. The product serves as a template for conventional PCR synthesis and detection is performed using short LNA probes.
Suppliers of real-time PCR thermocyclers
The basic methodology of real-time fluorescent detection involves the excitation of the fluorophores (typically using a lamp, light-emitting diode or laser) and emission detection using charge-coupled device cameras, photomultiplier tubes or other photodetectors. Fluorophore excitation and emission occur at wavelengths specific to the fluorophores used. Where broad spectrum excitation sources (e.g. lamps) are employed the excitation wavelength can be controlled by the use of filters. Similar technology is often used to detect only the desired emission wavelength by the detection source.
The suppliers of real-time PCR machines are numerous (e.g. Applied Biosystems, Bio-Rad, Roche, Stratagene and Cepheid) (Table 1). The main hardware-related differentiating factors between the various instruments are the throughput capability, assay well format, the number of wavelengths that can be scanned and PCR cycling rate. Most systems are designed to manage 96-well assay plates using conventional PCR cycling parameters on a heating block and are capable of detecting multiple wavelengths. This enables multiplexing of reactions that use differently labelled probe sequences. Variations on the common formats include the LightCycler system (Roche) that can perform a PCR in ~30 minutes through the use of assay capillaries with high surface area to volume ratios and heated air controlled thermocycling.The Cepheid SmartCycler allows the thermocycling characteristics of each reaction well to be controlled individually and their GeneXpert system fully automates the processes of DNA extraction and real-time PCR amplification. Recent advances have also seen the production of 384-well detection systems with the capability of performing standard PCR reactions in as little as 40 minutes (Applied Biosystems, 7900HT). This increase in throughput can be augmented with the use of robotic loading facilities and the microfluidics card. The microfluidics card can be purchased with selected assays pre-loaded into a 384-well plate. Using a system of micro channels, eight samples can be loaded into separate wells that are subsequently distributed through the channels into 48 different assay wells, thus eliminating the need to pipette the samples into each assay individually.

Pre-validated assays
The limiting time-related factor for many real-time PCR assays is the optimisation. Whilst this can be particularly prominent for SYBR green and multiplexed assays the availability of pre-validated ‘off the shelf’ assays has greatly reduced this burden. A number of repositories describe user-submitted PCR primers sequences and reaction conditions for assays that use all types of detection chemistry. Useful sources are the ‘Quantitative PCR Primer Database’, QPPD: http://web.ncifcrf.gov/rtp/gel/primerdb, ‘Primer Bank’7: http://pga.mgh.harvard.edu/primerbank, RTPrimerDB8: http://medgen.ugent.be/rtprimerdb and http://www.realtimeprimers.org.
Alternative resources are pre-validated assays using hybridisation probe chemistries that are available from commercial suppliers. The largest of these repositories involve the TaqMan detection chemistries by Applied Biosystems, which currently supply pre-validated kits for the detection of 400, 000+ human, mouse and rat transcripts with further assays available for other organisms.
Future directions and conclusions
The growing demand for higher throughputs of real-time PCR assays at an increasingly lower assay cost is exemplified by the introduction of 384 well assay formats, decreasing cycling time and robotic loading. Whilst technological improvements in optics and thermocycling capabilities are likely to progress these features still further, the use of advanced microfluidics identifies an area ripe for further research. One example is the Dynamic Array System that is currently in development and production by the Fluidigm Corporation. This reaction plate uses a series of microchannels and pressure controlled valves to direct the flow of assay reagents and samples into 10nl reaction chambers. The advantages of this format ensure that 96 samples and 96 assays can each be pipetted once into a plate and every assay-sample combination is derived within the reaction nano-chambers. Coupled with a fluorescent detection camera, this technology can perform 9216 (96 x 96) real-time PCR reactions in a single plate with minimal pipetting and minimal reaction volume thus enabling a reduction in assays costs.
Whilst the advancement of hardware technology is welcomed, this needs to be coupled with improvements in assay design, assay standardisation, data acquisition and data analysis. These developments should ensure that the potential of real-time PCR in the use of high-throughput and cost effective quantitative measurements is further realised. This will be particularly important as the requirement for validation of microarray data steadily increases and is likely to impact on many areas of research, diagnostics and commerce.
References
- Higuchi R, Dollinger G, Walsh PS, Griffith R: Simultaneous Amplification And Detection Of Specific DNA-Sequences. Bio-Technology 10(4), 413-417 (1992).
- Ginzinger DG: Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Experimental Hematology 30(6), 503-512 (2002).
- Williams NS, Gaynor RB, Scoggin S et al.: Identification and validation of genes involved in the pathogenesis of colorectal cancer using cDNA microarrays and RNA interference. Clinical Cancer Research 9(3), 931-946 (2003).
- Mackay IM, Arden KE, Nitsche A: Real-time PCR in virology. Nucleic Acids Research 30(6), 1292-1305 (2002).
- Hiratsuka M, Takekuma Y, Endo N et al.: Allele and genotype frequencies of CYP2B6 and CYP3A5 in the Japanese population. European Journal Of Clinical Pharmacology 58(6), 417-421 (2002).
- Bieche I, Olivi M, Champeme MH, Vidaud D, Lidereau R, Vidaud M: Novel approach to quantitative polymerase chain reaction using real-time detection: Application to the detection of gene amplification in breast cancer. International Journal Of Cancer 78(5), 661-666 (1998).
- Wang XW, Seed B: A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Research 31(24), (2003).
- Pattyn F, Speleman F, De Paepe A, Vandesompele J: RTPrimerDB: The Real-Time PCR primer and probe database. Nucleic Acids Research 31(1), 122-123 (2003).