

Proteomics and target identification in oncology
Posted: 16 February 2011 |
The recent progresses in the field of proteomics now enable large scale, high throughput, sensitive and quantitative protein analysis. Therefore, applying proteomics in clinical oncology becomes realistic. From the analysis of cell cultures to biological fluids and tumour biopsies, proteomic investigations of cancers are flourishing and new candidate biomarkers and therapeutic targets are slowly emerging. In the meantime, what we know of the cancer proteome is also an evolving figure that is progressively unveiled. Given the multiparametric nature and diversity of cancers, it should not be underestimated that a great deal of time and effort will be necessary for translating that knowledge into practical applications in oncology.
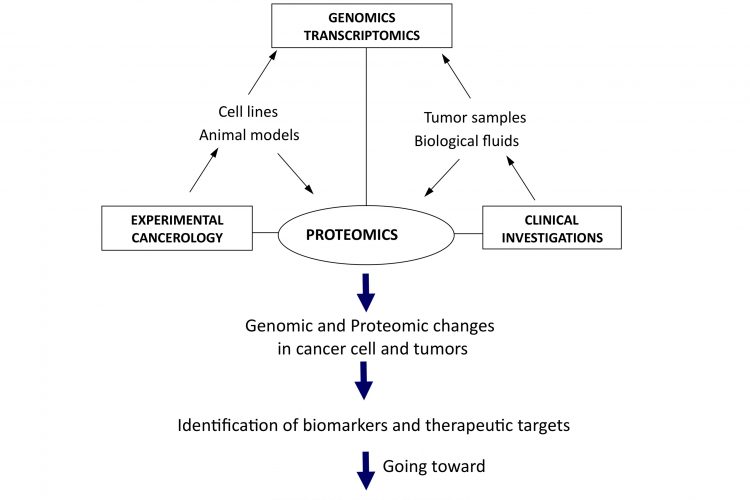
Figure 1Translational and integrative proteomics for the identification of new therapeutic targets in oncology
The recent progresses in the field of proteomics now enable large scale, high throughput, sensitive and quantitative protein analysis. Therefore, applying proteomics in clinical oncology becomes realistic. From the analysis of cell cultures to biological fluids and tumour biopsies, proteomic investigations of cancers are flourishing and new candidate biomarkers and therapeutic targets are slowly emerging. In the meantime, what we know of the cancer proteome is also an evolving figure that is progressively unveiled. Given the multiparametric nature and diversity of cancers, it should not be underestimated that a great deal of time and effort will be necessary for translating that knowledge into practical applications in oncology.
It is provocative, but nonetheless true, to state that in terms of biomedical applications, the major outcome of the human genome sequencing has been to open the way for the exploration of the proteome. With the progressive description of the human genome, it gradually became clear that looking solely at genes or mRNA would not be sufficient either for the full understanding of pathologies, or for the identification of definitive biomarkers and therapeutic targets. Beyond nucleic acids that are composed of only five bricks / nucleotides bearing rare modifications and are basically to store and transfer information, there is a huge level of complexity associated with proteins. Not only are proteins made of 20 major amino acids that can be subject to more than 200 types of post-translational modifications, but also proteins are actually making the structure and functioning of living entities by running most chemical reactions and metabolisms of other biochemical constituents, such as sugar or lipids, and even, ironically, nucleic acids. So here we are today, in the post-genomic era, trying to integrate genomics and proteomics to find new proteins, new post-translational modifications or new protein networks that could be of practical application to diagnose or treat diseases. The field of cancer makes no exception, and proteomics is expected to introduce a new dimension for the deciphering of the molecular mechanisms involved in tumour initiation and progression, with the identification of new therapeutic targets towards the goal of individualised / personalised treatments.
What’s new in proteomics and why it is now a reliable tool for identifying targets?
Since the invention of 2D electrophoresis in 1975, global methodological approaches to study the protein content of a biological sample have diversified and improved, but the turning point has certainly been the introduction of mass spectrometry. During the early 1990’s, the first identification of proteins extracted from a 2D gel by mass spectrometry has created such a synergy with progresses made in genome sequencing and in bioinformatics, that the word proteomics was coined just afterwards1. It is indeed important to remember that the three bases to the birth of proteomics were mass spectrometry / genome sequencing / bioinformatics. We perform proteomics the way we do it today because we can analyse proteins by mass spectrometry and because the genomes have been sequenced and data made searchable through bioinformatics. Importantly, this is true not only for protein identification, but also for characterisation of all post-translational modifications that rely on the same bases. Moreover, the sensitivity of mass spectrometry has constantly been improving and although we already reached the atomolar, it is anticipated, mainly due to miniaturisation and the input of nanotechnologies, that further increases in sensitivity are still to come2.
Interestingly, protein separation, which is crucial to proteomics, has also been improved, but with little contribution from mass spectrometry. Pre-fractionation and separation are indeed always needed. New technologies, like capillary electrophoresis or multi-dimensional chromatography, have been successfully introduced and even though they have not replaced 2D electrophoresis, they certainly add further capabilities of resolution, particularly for basic and low molecular weight proteins. They are also more appropriate for the analysis of a large number of samples as is very often the case with clinical investigations. As an illustration of the need for protein or peptide separation, mass spectrometers are now usually coupled with nano-chromatography in systems like LC-MS/MS or LTQ-FT mass spectrometer.
Aside from separation and sensitivity, the question of protein quantification has certainly been the most recently evolving aspect of proteomics, with several new approaches being introduced over the last few years. Traditionally protein quantification was performed on the entire protein, after electrophoresis or chromatography, using colorimetric or fluorimetric detection. From UV detection, comassie bue, silver nitrate or autoradiography after incorporation of radioactive amino acids, a range of approaches were available, but the quantification obtained were only relative and consisted more of a comparison of relative protein levels. Increase or decrease in individual proteins could be quantified, but a precise quantity – molarity – could hardly be obtained. Mass spectrometry by itself is not really quantitative and it is only with the introduction of stable isotope labelling that the situation has started to change3.
Methods like SILAC (Stable Isotope Labelling in Cell Culture), based on the incorporation of labelled amino acids with different molecular weights, enables mass spectrometry protein quantification based on the quantification of several trypsin generated peptides. When working with biopsies, where incorporation of modified amino acids is not feasible, chemical labelling of peptides can also allow mass spectrometry based quantification, as illustrated with iTRAQ (isobaric Tag for Relative and Absolute Quantitation). Finally, what is often considered as the current best way to provide a true quantification of a peptide in a sample is the AQUATM system (Absolute QUAtification, Sigma), in which a synthetic peptide, labelled with stable isotope and corresponding to a defined protein, is used as an internal standard for quantification in mass spectrometry4.
Interestingly, for explorative investigation, the precision of modern mass spectrometers such as FT-ICR or ORBITRAPTM plus advanced bioinformatic analysis has rendered possible the concept of ‘label free’ quantification. In this approach, no labelling at all is used and the levels of different peptides from a defined protein are merged to give an evaluation of the quantity5. To be clear, it is not matching the precision obtained with SILAC, iTRAQ or AQUA, but it gives an order of magnitude that can be of value, particularly in a clinical context where the other methods can eventually be challenging to implement. In conclusion, the recent years have seen the introduction of a range of practical approaches that render proteomics really quantitative, and in terms of biomarker and target identification, it is certainly making a considerable difference.
It is a fact that until very recently, most research to identify targets in proteomics has been performed using differential analysis that did not involve very reliable quantification methods, and as a consequence, a number of potential targets that were identified in early stages turned out to be false positive during validation phases. In this context, the development of Multiple Reaction Monitoring (MRM) is certainly a milestone for validation of potential targets and biomarkers, as precise quantification of a set of peptides, corresponding to one or several proteins, can be established in a way that is fully compatible with large scale analysis of many samples6. This is illustrated by a pertinent study that has looked at PSA (Prostate Specific Antigen) in prostate cancer, using MRM and demonstrating its potential for assaying cancer related proteins7. Antibody arrays should also be mentioned, although their practical use has not yet reached the same level of intensity, the repertoire of antibodies that have highly selective binding to native or post-translationally modified protein epitopes has expanded rapidly. This is now increasingly developed and many companies offer commercial and customised arrays8. Together, just as observed with DNA microarrays, the lack of precise quantification was a very limiting factor to proteomics based investigation, but this limitation is now being overturned, hence creating a more secure environment for target identification.
Implementing proteomics for target identification in oncology
These recent technological advances are now changing the perspectives for application in the field of cancer9, and one of the best illustrations is provided by the use of proteomics to dissect tyrosine kinase signalling pathways. Tyrosine kinase receptors and associated signalling networks play a crucial role in cancer initiation and progression, and some of them, such as the variant EGF receptor Erb-B2, are already targeted by anti-cancer treatments like Herceptin or specific anti-kinase inhibitors. Proteomics can be used to decipher the molecular cascades initiated by tyrosine kinase receptors and even the kinetics of kinase activation and deactivation can be investigated, along with the activity of specific inhibitors of potential therapeutic value10. EGFR, the most investigated tyrosine kinase receptor in cancer cells, has naturally been studied, but more rare receptors have also been analysed with proteomics, for instance, the proto-oncogenic tyrosine kinase receptor TrkA, which is a receptor for nerve growth factor and stimulates proliferation and invasion of breast cancer cells11.
A proteomics-based study has evidenced a series of TrkA signalling partners that are related to cell growth and, interestingly, the DNA repair protein Ku70 was identified as involved in TrkA signalling12. It was shown that during the course of stimulating cell proliferation, TrkA induces the activation of Ku70 to prevent apoptosis, reinforcing the aggressiveness of breast cancer cells and their progression towards a more advanced cancer phenotype. Those data point to trkA and Ku70 as potential new targets in breast cancer. Another example is given with the serine / threonine kinase AKT in breast cancer. AKT is crucial to most cancer development, as its impairment inevitably results in apoptosis of cancer cells, placing AKT as a potential therapeutic target13. However, AKT is a largely distributed kinase which also controls a variety of other cellular processes, such as metabolism, and therefore targeting AKT is likely to have a wide series of side effects, limiting practical applications. A way to overcome this problem would be to define signalling proteins downstream to AKT and specifically involved in tumour cell survival. Proteomics exploration has revealed a list of potential AKT interacting partners that are related to cell survival14. Among them, and of particular interest, was the ATPase valosin-containing protein (VCP) which appears to be required for AKT anti-apoptotic signalling. VCP interacts and is a substrate of the kinase activity of AKT, the serine residues involved have been identified and their mutation resulted in a potent induction of cancer cell apoptosis15.
Importantly, the same study reported that the impairment of AKT/VCP interaction potentiated the effect of several chemo – therapeutic drugs, such as 5FU or etoposide, indicating the potential interest of targeting VCP in breast cancer therapy. In addition to helping in the deciphering of signalling pathways, proteomics can also provide structural information on oncoproteins or suppressor proteins that could lead to future potential therapeutic strategies. For example, the oncosuppressor p53, for which a full sequencing and determination of posttranslational modifications has been performed by mass spectrometry FT-ICR16. The data reveal a pattern of phosphorylation and acetylation in the different domain of the protein, confirming previous data obtained with antibodies, and highlighting new acetylation sites that provide new opportunities not only for the better understanding of p53 activation, but also for future targeting.
Despite significant outcomes of proteomics for target identification, it has to be acknowledged that none of the targets identified so far by proteomics have actually entered into clinical practice in oncology. However, this should not be such a disappointment, not only because a mean of 10 years is usually necessary between discovery and clinical use of both biomarkers and therapeutic targets, but also because we have so far only explored the most obvious aspects of the human proteome. Despite a lot of agitation and emphasis around proteomics and its potential in biomedicine, one should not forget that most of the complexity of the proteome, in terms of protein numbers and post-translational modifications, is still not accessible with our current tools. With about a million proteins and their post-translational modifications, we have explored so far in cancer – as in other pathologies – only the surface of it and we are going to need much more time and efforts, along with technological improvements, to have access to the deep proteome. More than 30 years after Nixon’s declaration of war against cancer, we are still facing this multifactorial and diverse disease which is constantly progressing. It is a fact that no major breakthrough (like the discovery of antibiotics for infectious disease) has been made and we are now entrenched in what looks like a long battle against cancer. Although it is too early to know if proteomics will change the course of this war, it is clearly a promising weapon introduced in the battlefield.
The next step: integrating proteomics into a bigger picture
Now that more and more data are being collected about the proteome of different cancers, from cell culture, biopsies or biological fluids, a concomitant need to have a better integration appears. Just as information about the sole genome does not allow comprehension of complex biological systems, a proteomic view alone is equally not sufficient. If proteins are the functional outcomes of genes, they are not the end point of gene transcription, and ultimately, proteins only drive cells towards appropriate phenotypes and behaviours; therefore further understanding of complex cancer systems now requires more integrated approaches and strategies.
The need for a better integration can be regarded at two different levels. Firstly from the molecular point of view, putting together genomics, transcriptomics and proteomics data would give access to a more global view of molecular changes occurring in cells during the initiation and progression of cancer, as well as potential ways for rational intervention to destroy or normalise them. The second possible level of integration is from a more functional perspective, taking into consideration the cellular interactions occurring in most cancers, at least in solid tumours, where there are many interactions between cancer and surrounding normal cells of different origin, like fibroblasts or endothelial and immunocompetent cells. From the study of xenografted human cancer cells in immunodeficient mice to the use of isogenic models, animal models also offer the possibility to integrate data with genomic analysis for a better knowledge of global in vivo molecular modifications associated with tumorigenesis. Together, it seems clear that what could be called systems proteomics, with integration at both the molecular and physiological levels, is under way to open a new dimension for both differential and functional analyses of the cancer proteome (Figure 1). That is definitely contributing to the evolution of the field towards personalised management and treatment of cancer. In this new era, initiatives like the Human Protein Atlas (http://www.proteinatlas.org/) or the worldwide organisation HUPO (Human Proteome Organisation, http://www.hupo.org/) are particularly important to bring together expertise and to provide the framework and integrative programmes aimed to future identification and validation of therapeutic targets in oncology.

Figure 1Translational and integrative proteomics for the identification of new therapeutic targets in oncology
Acknowledgements
This work was supported by the University of Lille, the French National Institute of Health (INSERM) and the ‘Ligue Nationale Contre le Cancer’ (Equipe Labellisée 2009).
References
1. Kahn P. From genome to proteome: looking at a cell’s proteins. Science. 1995 270:369-70
2. Gstaiger M, Aebersold R. Applying mass spectrometrybased proteomics to genetics, genomics and network biology. Nat Rev Genet. 2009 10:617-27
3. Gevaert K, Impens F, Ghesquière B, Van Damme P, Lambrechts A, Vandekerckhove J. Stable isotopic labeling in proteomics. Proteomics. 2008 8:4873-85
4. Brun V, Masselon C, Garin J, Dupuis A. Isotope dilution strategies for absolute quantitative proteomics. J Proteomics. 2009 72:740-9
5. Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol. 2010 2010:840518
6. James A, Jorgensen C. Basic design of MRM assays for peptide quantification. Methods Mol Biol. 2010 658:167-85
7. Fortin T, Salvador A, Charrier JP, Lenz C, Lacoux X, Morla A, Choquet-Kastylevsky G, Lemoine J. Clinical quantitation of prostate-specific antigen biomarker in the low nanogram/milliliter range by conventional bore liquid chromatography-tandem mass spectrometry (multiple reaction monitoring) coupling and correlation with ELISA tests. Mol Cell Proteomics. 2009 8:1006-15
8. Sanchez-Carbayo M. Antibody array-based technologies for cancer protein profiling and functional proteomic analyses using serum and tissue specimens.Tumour Biol. 2010 31:103-12
9. Collins BC, Lau TY, O’Connor DP, Hondermarck H. Cancer proteomics-an evolving battlefield. Conference on Cancer Proteomics 2009: Mechanistic Insights, Technological Advances & Molecular Medicine. EMBO Rep. 2009 10:1202-5
10. Kolch W, Pitt A. Functional proteomics to dissect tyrosine kinase signalling pathways in cancer. Nat Rev Cancer. 2010 9:618-29
11. Dolle L, Adriaenssens E, El Yazidi-Belkoura I, Le Bourhis X, Nurcombe V, Hondermarck H. Nerve growth factor receptors and signaling in breast cancer. Curr Cancer Drug Targets. 2004 4(6):463-70
12. Com, E., Lagadec, C., Page, A., El Yazidi-Belkoura, I., Slomianny, C., Spencer, A., Hammache, D., Rudkin, B.B., Hondermarck, H. Nerve Growth Factor Receptor TrkA Signaling in Breast Cancer Cells Involves Ku70 to Prevent Apoptosis. Mol. Cell. Proteomics. 2007, 6, 1842-1854
13. Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des. 2010 Jan;16(1):34-44
14. Vandermoere, F., El Yazidi-Belkoura, I., Demont, Y., Slomianny, C., Antol, J., Lemoine, J., Hondermarck, H. Proteomic exploration reveals that actin is a signaling target of the kinase akt. Mol. Cell. Proteomics 2007, 6, 114-124
15. Vandermoere, F., El Yazidi-Belkoura, I., Slomianny, C., Demont, Y, Bidaux, G., Adriaenssens, E., Lemoine, J., Hondermarck, H. The valosin-containing protein (VCP) is a target of AKT signaling required for cell survival. J. Biol. Chem. 2006, 281, 14307-13
16. Joubel A, Chalkley RJ, Medzihradszky KF, Hondermarck H, Burlingame AL. Identification of new p53 acetylation sites in COS-1 cells. Mol Cell Proteomics, 2009, 8:1167-73
About the Author
Dr. Hubert Hondermarck is Professor of molecular and cellular biology at the University of Lille, member of the Institut Universitaire de France and head of the U 908 INSERM (French National Institute of Health) laboratory working on growth factor signalling in breast cancer. The aim of his work is the deciphering of cancer cell signalling in the context of tumour growth and metastasis, for the identification of new biomarkers and therapeutic targets in oncology. He has been President of the French proteomics society (SFEAP) and is a member of the board of Directors of the Human Proteome Organisation (HUPO).
