Recent developments in the use of LCMS in process pharmaceutical chemistry
Posted: 28 February 2012 |
Liquid Chromatography Mass Spectrometry (LCMS) is a powerful technique that has recently undergone exponential growth in its application to pharmaceutical synthesis. This perspective will outline the general principles of LCMS, detail some recent approaches and the benefits to be derived from its use at an early stage of process development. Identification of the components in a mixture is the primary function of analytical chemistry and there are a range of techniques available. When the solution to this problem requires some structural identity, LCMS is the instrument of choice. Liquid Chromatography Mass Spectrometry is defined as the use of the separating properties of liquid chromatography combined with a detector capable of mass analysis (mass spectrometer: single quadrupole, triple quadrupole, ion trap, Time Of Flight, Q-TOF etc.). This combination may be configured in many ways, for a general scheme…
Liquid Chromatography Mass Spectrometry (LCMS) is a powerful technique that has recently undergone exponential growth in its application to pharmaceutical synthesis. This perspective will outline the general principles of LCMS, detail some recent approaches and the benefits to be derived from its use at an early stage of process development.
Identification of the components in a mixture is the primary function of analytical chemistry and there are a range of techniques available. When the solution to this problem requires some structural identity, LCMS is the instrument of choice.
Liquid Chromatography Mass Spectrometry is defined as the use of the separating properties of liquid chromatography combined with a detector capable of mass analysis (mass spectrometer: single quadrupole, triple quadrupole, ion trap, Time Of Flight, Q-TOF etc.). This combination may be configured in many ways, for a general scheme.
In principle, the mixture of components to be separated is loaded on an autosampler, injected into an LC stream of mobile phase and separated on a column. The eluting fractions are then detected by a mass analyser (or commonly an in-line UV/DAD detector followed by a mass analyser) via ionisation in one of a number of techniques (EI, ESI, APCI, APPI, ESCI etc.). With respect to the LC setup, this can be HPLC or UPLC depending on the analytical method and there are obvious merits for each. There are a multitude of options on the characteristics of the mass analyser.
Mass spectrometry hardware
The variety of mass analysers capable of LCMS operation can be defined as the method by which mass is differentiated and by the experiment to be undertaken. In brief, a single quadrupole detector is commonly used for simple mass analysis; a triple quadrupole/ion trap for fragmentation analysis; a Time of Flight (TOF) for high resolution measurements and a Q-TOF for high resolution measurements of fragmentation patterns.
Simple mass analysis will give molecular ion data to the integer level and is very useful as a preliminary screen to see if the sample will ionise under the operating conditions. This is commonly undertaken to confirm the presence of an ion in a sample before looking at more elaborate techniques or is used in quantification. Fragmentation analysis is undertaken in order to aid identification of components within mixtures that separate in LC conditions. The data revealed by this process identifies the smaller fragments that when bonded together form the molecular ion seen in simple mass analysis. The use of a triple quadrupole or an ion trap instrument is associated with fragmentation experiments as there are two quadrupoles either side of a collision cell. The collision cell is where the ion fragmentation occurs but the ion beam can be scanning or selective before or after the collision cell. This essentially means the triple quadrupole is capable of scanning for molecular ions/fragments or selective monitoring of individual molecular ions/ fragments. This is a very powerful technique as it can be used as a key determinant in the assessment of structure and again can be used for quantification.
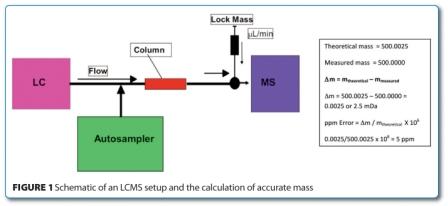
The addition of a reference standard (or lock mass) and the use of a high resolution detector allows acquisition of mass to four decimal places and hence for elemental composition to be measured. This can be compared to that calculated and is a very common technique in the identification of unknowns. As a guiding principle this must fit to within 5ppm given the formula in Figure 1, above (though this is highly dependent on theoretical mass) and the technique works best if the sample history is well known as this will limit the potential parameters.
The significant advantage of LCMS over other liquid chromatography detectors is the quality of information on detection of a peak and as such is commonly used as one of a suite of techniques to determine structural information on a component.
The use of LCMS in pharmaceutical process development
The use of LCMS in a pharmaceutical setting is well established as analysts have always sought the best quality information from detectors and the outlay involved in set-up has significantly decreased. The seeding point for much of what is described below has been in the industrial QA/QC laboratories (due to the obvious qualitative and quantitative data produced) but also in academic collaborations. Recently, the application of LCMS (in tandem with other qualitative and theoretical techniques) to process development has gained significant momentum. Outlined below are some recent developments in the use of LCMS applied to process development change, a very significant area in pharmaceutical chemistry having achieved global investment in the past decade.
Pharmaceutical applications of LCMS
In a recent publication from AstraZeneca, the process synthesis of a potent SRC kinase inhibitor (AZD0530) was revealed with the use of LCMS and molecular modelling key to the development. The preliminary synthetic route involved three successive nucleophilic aromatic substitutions and the key focus of this work was the final SnAr reaction which gave significant levels of by-product formation as identified by HPLC (and relatively poor isolated yield of only 63 per cent). Initial studies identified that by a change of base and solvent mixture from sodium t-amylate and t-amyl alcohol to sodium hydroxide and toluene resulted in a significant improvement in reaction profile. To test the hypothesis, the introduction of t-amyl alcohol to the toluene route significantly deteriorated the LCMS profile of the reactions with more hydrolysis products evident.

As can be seen in Scheme 1, this final step was optimised to now yield <13 per cent impurities and had significant advantages over the initial method: toluene allowed for higher reaction temperatures and reaction time and alcohol equivalents were halved1.
In another recent publication, chemists at Sanofi disclosed the process development of a Casein Kinase I – Epsilon inhibitor 3-(3-fluorophenyl)sulfanyl-1H-pyrrolo[3,2-b] pyridine-2-carboxylic acid amide2. The discovery route involved a number of steps which suffered from moderate yields and poor reproducibility requiring optimisation. LCMS was utilised to track the intermediates and impurities of a key hydrogenation/cyclisation of a nitropyridine to a 4-azaindole (as can be seen in Scheme 2 (below), the postulated impurities were hydroxylamines and 1-hydroxy-5-azaindoles). Catalytic hydrogenation from the first gen – eration route was replaced by transfer hydrogenation on scale-up with the purity of ketoester starting material as well as concentration of ammonium formate proving the key to success.

LCMS has also been used to track the progress of an asymmetric enzymatic resolution in research by a group from Eli Lilly looking at the synthesis of enantiomerically pure β-prolines3. Initially, the hydrolysis of a series of β-proline methyl esters by 13 hydrolytic enzymes were determined by achiral LCMS (conversions of about 50 per cent) and their optical purity by chiral GC with up to 96%ee’s recorded. Consequently, the reverse esterification reaction was screened with a series of lipases from the racemic carboxylic acid showing low selectivity at first attempts. Development of a chiral LCMS method (using a CHIRALPAK AD column) gave baseline separation of the four components (both enantiomers of starting ester and acid) and allowed for automation of the chemistry and the analysis. After a total optimisation of the process led by the chiral LCMS data, the procedure in Scheme 3 (below) was adopted to yield both enantiomers of N-Boc protected β-proline in high enantiomeric excess.

In another example of the use of LCMS by process chemists the development of a chiral epoxide precursor to a CCR1 antagonist was reported by AstraZeneca4. The original medicinal chemistry route to this key intermediate derived from a Sharpless asymmetric epoxidation followed by nosylation and displacement with a phenol. The intermediate nosylate was unsuitable for scale-up and novel chemistry was devised to obviate this intermediate. After much process development, a route through a protected diol to form the epoxide in the final step was chosen with LCMS as well as ReactIR, near IR and NMR aiding the development process. The use of a carefully controlled temperature process significantly reduced the production of oligomeric by-products of the phenoxide and led to multikilogram compatible chemistry.
Another process development, this time in the area of cancer chemotherapy, has been published by GSK in the synthesis of a scalable route to GSK923295A5. GSK923295A is selective inhibitor of CENP-E motor domain enzyme activity which has exclusive functions in mitosis. Starting from readily available phenylalaninol, a Friedel-Crafts acylation (favouring the para product) and derivatisation to a pyridyl imidazole was postulated to yield the target compound. However, on heterocycle formation, a novel by-product was identified by LCMS and fully confirmed by de novo synthesis. The use of TBAB appears to significantly enhance the reaction rate but also gives rise to the brominated by-product as seen in Scheme 5, below. Indeed the potential for bromination of this imidazole was borne out by a test reaction where the instantaneous and quantitative conversion of GSK923295A intermediate to the by-product on addition of NBS was seen. The reactivity of imidazoles towards bromination is well documented but the source of electrophilic bromine in this case is yet to be identified.

The synthesis of a novel CRF1 receptor antagonist, recently described by Bristol-Myers Squibb, also contains process developments led by LCMS. One of the key intermediates in this synthetic route is the substituted aminopyrazole seen in Scheme 6 (below) which is converted to a pyrazolotriazine.

On initial aminopyrazole formation, a by-product (up to 10 per cent) was identified by HPLC and LCMS analysis identified that it had the same mass as the required imidate suggesting an isomer. In order to confirm this, the aminopyrazole was heated in toluene at 100°C which resulted in a 90:10 mixture of aminopyrazole and isomer. Subsequent purification and NMR analysis assigned conclusively the structure as the acyl migration product found to exist in equilibrium. After a series of screening experiments on the acid catalyst, it was seen that the tosic acid should be replaced by trifluoroacetic acid to yield the final process.
Another pharmaceutical process benefiting from the use of LCMS is the recently disclosed synthesis of chiral 2-morpholines which appear in a number of pharmacologically active compounds such as reboxetine. The typical process impurities identified can be seen in Scheme 7 – with the key enantiomeric impurity being the major concern.

After a screen of recrystallisation solvents, the use of 2-propanol at high temperatures was identified to have a significant impact on the levels of the enantiomeric impurity. Indeed, testing this hypothesis by heating a solution of the chiral morpholine (with an ee of >98 per cent) in 2-propanol and measuring the enantiomeric excess (ee%) a drop was seen after eight hours (97.5 per cent) and even further after 24 hours (80 per cent). This discovery led to significant improvements in process yield. The other key impurity was identified by LCMS in certain lots of chiral morpholine as an isobutyl ketone and was ratified by independent synthesis. After a number of trial experiments to identify the cause of this impurity, it appears to be due to the use of DIBAL as an initiator in the Grignard reaction to produce the chiral morpholine.
Conclusions
As can be seen in key examples from the past couple of years, LCMS in pharmaceutical process development is fast becoming an essential technique. The ability to identify the com ponents of a process reaction mixture has led to significant improvements in yields, impurity profiles and reactor turnaround and is set to be a mainstay in pharmaceutical institutions.
About the author
Florence McCarthy graduated from the School of Pharmacy, University of Sunderland, UK. In 2001, Dr. McCarthy joined the Auckland Cancer Society Research Centre (ACSRC), University of Auckland, New Zealand in conjunction with Pfizer Global Research and Development working on inhibitors of the Wee1 and Chk1 checkpoint kinases and the ErbB kinase inhibitor programme. On his return to Ireland, he joined UCC as a parttime Lecturer in the School of Pharmacy and the Department of Chemistry in 2003 before being made permanent in the Department of Chemistry in 2005. Currently, Dr. McCarthy leads a team of researchers in medicinal and pharmaceutical chemistry investigating the synthesis and evaluation of diverse bioactive molecules from steroids to complex heterocycles. He is a Principal Investigator in the Analytical and Biological Chemistry Research Facility (ABCRF) and also runs the ABCRF mass spectrometry laboratory providing LCMS services to most of the multinational pharmaceutical corporations in Ireland, as well as within UCC and to many other third level institutions.
References
1. A Simplified Process for the Manufacture of AZD0530, a Potent SRC Kinase Inhibitor; Steven A. Raw, Brian A. Taylor, and Simone Tomasi; Org Proc Res Dev 2011, 15, 688-692
2. Chemical Development of the Casein Kinase I – Epsilon Inhibitor: 3-(3-Fluorophenyl)sulfanyl-1Hpyrrolo[ 3,2-b]pyridine-2-carboxylic Acid Amide; Bao- Guo Huang, Gregory Kubiak, John J. Shay, Clive Pemberton, James Peers, Reda G. Hanna, Matthew R. Powers, Juan A. Gamboa, Ann M. Gelormini, Hyacinthe Yarabe, and Duane E. Rudisill; Org. Process Res. Dev. 2011, 15, 1040–1045
3. Enzymatic Resolution of N-Substituted-β-prolines; Javier Mendiola, Susana García-Cerrada,, Óscar de Frutos, María Luz de la Puente, Rui Lin Gu, and Vien V. Khau‡ Organic Process Research & Development 2009, 13, 292–296
4. Development of a Multikilogram Synthesis of a Chiral Epoxide Precursor to a CCR1 Antagonist. Use of in Situ Monitoring for Informed Optimisation via Fragile Intermediates; Debra Ainge, James E. M. Booker,† Nicholas Pedge, Rhona Sinclair, Chris Sleigh, Marijan Sˇ tefinovic´, Luis-Manuel Vaz, and Edward Way. Organic Process Research & Development 2010, 14, 72–84
5. Discovery and Development of an Efficient, Scalable, and Robust Route to the Novel CENP-E Inhibitor GSK923295A; Richard Bellingham, A. Mark Buswell, Bernie M. Choudary, Andrew H. Gordon,, Steve O. Moore, Matthew Peterson, Mike Sasse, Amin Shamji, and Michael W. J. Urquhart. Organic Process Research & Development 2010, 14, 1254–1263
6. The Development of a Robust Process for a CRF1 Receptor Antagonist; Sevrine Broxer, Monica A. Fitzgerald, Chris Sfouggatakis, Jessica L. Defreese, Evan Barlow, Gerald L. Powers,Michael Peddicord, Bao-Ning Su, Yue Tai-Yuen, Charles Pathirana, and James P. Sherbine; Org. Process Res. Dev. 2011, 15, 343–352
7. Practical Synthesis of Chiral 2-Morpholine: (4-Benzylmorpholin-2-(S)-yl)-(tetrahydropyran-4- yl)methanone Mesylate, a Useful Pharmaceutical Intermediate. Michael E. Kopach, Utpal K. Singh, Michael E. Kobierski, William G. Trankle, Michael M. Murray, Mark A. Pietz,Mindy B. Forst, and Gregory A. Stephenson, Vincent Mancuso, Thierry Giard, Michel Vanmarsenille and Thierry DeFrance; Organic Process Research & Development 2009, 13, 209–224
Issue
Related topics
Clinical Development, Liquid Chromatography - Mass Spectrometry (LC-MS), Mass Spectrometry