

Pharmaceutical proteomics: a journey from discovery and characterisation of targets to development of high-throughput assays
Posted: 15 December 2013 |
Proteomics has evolved during the last few years from a time-intensive, cost-intensive and hard-to-reproduce technique in basic research to a versatile and reliable tool in various areas of pharmaceutical research. The exploding progress in mass-spectrometry-compatible protein and peptide-separation methods led to the development of new approaches particularly suited for monitoring a multitude of specific targets in highly complex matrices in a highly sensitive, specific and parallel fashion. These new technologies have caused a paradigm shift in proteomics from mostly gel-based, hypothesis-generating studies towards fast, cost-effective and mostly LC-MS-based assays. Therefore, proteomics emerges for pharmaceutical researchers aiming to identify and verify proteinaceous biomarkers as proteomics technology comes of age.
This mini-review highlights some of the recent technical developments that facilitate the use of proteomics as a tool for important steps in target protein identification, characterisation, biomarker discovery, validation and monitoring and discuss advantages and disadvantages of individual techniques.
Proteomics: from past to present
The term ‘proteome’ was coined as the entirety of all expressed proteins encoded in a genome1. In contrast to the genome which remains rather unchanged during an organisms` life and is the same for every cell, the proteome is not only altered by cell-type but is also highly dynamic due to various intrinsic and extrinsic factors that may affect it even within a few seconds. Proteomics therefore aims at a quantitative description of all proteins of a cell in parallel and their regulations by different stimuli. The necessity to identify and quantify thousands of proteins in parallel, typically from rather tiny available amounts of sample, poses a great challenge to analytical methods.
Protein analytical techniques used in proteomics nowadays like gel-based protein separations in one or two dimensions and chromatographic techniques for separation of peptides have been known and widely used for decades. Yet, protein identification e.g. by Edman-sequencing was restricted to pure proteins in rather large amounts and was therefore very time-consuming and often not successful. While protein and peptide separation techniques were continuously improved in separation power, sensitivity and dynamic range, protein identification remained the bottleneck in protein analysis and prevented their analysis on a proteomic scale.
The evolution of proteomics from protein analysis really gained momentum with the availability of genomic databases from the genome era and the development of soft ionisation techniques enabling the analysis of peptides and proteins by (tandem-) mass spectrometry. Electrospray ionisation (ESI) particularly became a widespread technique because it facilitates the direct coupling of liquid chromatography to tandem mass spectrometry, thereby allowing reliable high-throughput protein identification also from high-complexity mixtures.
Development and continuous improvement of the core techniques of proteomics, most notably protein electrophoresis, peptide chromatography and tandem mass spectrometry, paved the way for proteomics as a versatile tool in all stages of pharmaceutical research.
Analysis of intact proteins: top-down analyses
The mass spectrometric measurement of intact proteins has the benefit of reflecting the overall modification state of the respective protein and recent developments in mass spectrometric instrumentation also facilitates top-down sequencing of proteins2. By these means, the proteins can not only be identified but modifications on the proteins such as acetylations or phosphorylations can be localised to individual amino acid residues. These analyses are typically carried out on high-resolution mass spectrometers with FT-ICR-, orbitrap- or QTOF-analysers that feature the necessary mass accuracy and resolution for the analysis of the highly-charged analyte ions. The major limitations of the top-down mass spectrometry-techniques still lie in the insufficiency in the analysis of high-complexity mixtures and the fragmentation of large proteins as MS/MS-spectrum complexity rises tremendously with protein size and full sequence coverage is typically not obtained above 20 kDa.
While top-down mass spectrometry has not yet been established as a standard method in exploratory proteomics studies, it is widely used as a tool for quality control, particularly in the characterisation of antibodies. The possibility to distinguish different isoforms and monitor the modification state and purity of therapeutic antibodies makes this technique an invaluable tool in the pharmaceutical industry3.
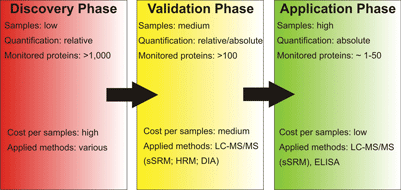
Protein biomarker studies can be divided into three phases being the identification of possible biomarker candidates (discovery phase), validation or refusal of the potential candidates (validation phase) and the monitoring of the identified biomarkers in a clinical setting (application phase). Mass spectrometry-based proteomics can aid in all three phases of this process using different techniques tailor-made for the task at hand
Analysis of protein interactions
Protein interactions fulfil essential functions in various different cellular processes. As the majority of proteins carry out their functions as protein complex constituents or in bilateral interactions in either a stable or transient fashion, characterisation of the interaction networks is a field in proteomics research with a longstanding record4,5.
The analysis of the ‘complexome’ in its entirety is a highly challenging and presumably impossible task for contemporary proteomics. Nevertheless, several studies applying mostly native gel-electrophoresis techniques for the separation with subsequent bottom-up mass spectrometry for protein identification have been carried out6-8. Also, differential complex analysis using DIGE in a native electrophoresis setting have been reported9,10. Top-down mass spectrometric measurements of protein interactions or protein complexes11 are scarce since the necessary techniques have been established only very recently and still have to prove their superior performance to bottom-up mass spectrometry approaches.
Another possibility for the analysis of protein interactions from in vivo samples is co-immunoprecipitation using a specific antibody for the chosen bait protein as subsequent identification of the isolated proteins is typically no big challenge nowadays. However, specificity of the co-immunoprecipitation is crucial as the major challenge is distinction of true interactors of the bait protein and contaminating proteins. Quantitative proteomic approaches have been undertaken to sort out the contaminations, but still transient or low-affinity interactors might be concealed by higher abundant, unspecifically co-isolated proteins. Therefore, an increased amount of work has to be invested into subsequent validation of potential interactor candidates.
Co-immunoprecipitations are almost impossible to implement on a global scale due to these analytical limitations, particularly the lacking availability of suitable antibodies. Therefore, a widely used alternative is overexpression of tagged versions of the proteins of interest that can subsequently be used for tag-based isolation of the bait and its interacting partners. While these approaches can be accomplished in high-throughput, they are prone to false-positive identifications due to proteins that either interact with the tagging moiety or are co-purified as they have affinity for the tag-binding support, e.g. proteins with oligo-histidine-strands like YY1 that are co-purified by Ni2+-based IMAC12. Thus, the choice of the tag is a crucial point in such analyses.
Novel, multi-step tagging procedures can also be used to reduce the amount of contaminating proteins. Thereby, two affinity tags are attached to the bait protein with a protease cleavage site between the two tags. The bait and interacting proteins are affinity captured in a first step and released specifically by protease treatment. The second affinity purification using the now available second tag yields much purer protein complexes in comparison to one-step procedures although the amounts of isolated protein are typically lower13.
Modificomics: analysing protein modifications
The detection and localisation of protein modifications is of highinterest in many proteomics studies as they regulate various protein functions ranging from protein localisation over interactions and enzymatic activity to signal transduction events. Particularly phosphoproteomics has gained enormous popularity during the last decade14 as protein phosphorylations often act as fast molecular switches regulating protein activities and reliable, high-throughput-capable enrichment methods for phosphoproteins and peptides have become available. While nowadays up to a few thousand phosphorylation sites can be identified from a sample, there is still a need for quantitative data in order to understand the highly dynamic relations and regulations of individual phosphorylation sites and entire phosphorylation cascades. As enrichment of phosphoproteins and/or peptides is not easily established in a quantitatively reproducible manner, stable-isotope labelling techniques may be more straightforward for quantitative phosphoproteomics than label-free techniques.
The variety and complexity of protein modifications beyond phosphorylation like glycosylations, acetylations, ubiquitinylations and more than 600 other known modifications are far too large to be considered here and are specifically addressed by further reviews15-17.
The need for spatio-temporal proteomics in systems biology
Contemporary proteomics data to be used in systems biological studies not only has to be generated in a quantitative fashion, but also with spatial and / or temporal resolution18,19. The high dynamics in cellular processes raise the need for the study of entire courses of action rather than the comparison of two distinct states at an often arbitrarily chosen point of time after disease onset or initiation of a treatment. Analyses lacking a temporal aspect are very likely to miss the important molecular processes underlying cellular reactions or may – even more severe – lead to false conclusions. These considerations equally apply to spatial resolution as protein translocation events like recruitment of small G-proteins to membranes, nuclear import of transcription factors or even cytochrome c-release from mitochondria are events of utmost importance in the cell that can not be detected by the analysis of whole cell lysates, e.g. by shotgun-proteomics approaches. Despite the enormous effort in reproducible subcellular fractionation for various cellular compartments at several points in time, aiming for spatio-temporal resolution also increases the number of samples to be analysed tremendously. Therefore, stable-isotope labelling-based approaches are not very well suited for such studies merely due to the high cost of the labels and label-free approaches – although making much higher demands on the technology in terms of accuracy and reproducibility – will become the standard tools in systems biological studies.
Proteomics in biomarker discovery: proteomics in the identification phase
Identification of potential proteinaceous biomarkers from highly complex samples is the first and presumably the hardest task in biomarker-focussed proteome research (see Figure 1). Samples may originate from various different sources and typically studies are aimed at identification and quantification of as many proteins as possible including different isoforms. Therefore, sample preparation not only has to be customised to the sample at hand, but it must also be compatible to a variety of sample separation and identification methods as a comprehensive view of a proteome can only be achieved by combination of complementary analysis techniques. The variety of analysis approaches used for identification and quantification on the ‘entire proteome-level’ thus ranges from classical gel-based methods like 2D-PAGE and GeLC-MS to so-called ‘shotgun-proteomics’ and protein antibody arrays. Hence, using differential proteomics as a tool for biomarker discovery already requires a large arsenal of methods. However, differential proteomics comparing a ‘healthy control’ to a ‘diseased sample’ is only capable of identifying differentially regulated proteins between these two samples, but a good biomarker protein has many more prerequisites to match than being regulated between healthy and diseased state. For instance, the regulation has to specific for the respective disease; it has to be more pronounced than inter-individual alterations; the biomarker has to be stable, easy to obtain (e.g. from blood or urine) and detectable in an easy, fast and cost-efficient way. Therefore, further validation steps and efficient monitoring techniques have to be established; and proteomics techniques can also aid in these areas of pharmaceutical research.
Proteomics in the validation phase
When a set of potential biomarkers has been identified, it has to be validated typically in a larger number of patients by various independent methods. One of these methods can be mass spectrometry-based proteomics23. Usually from this point on, the applied proteomics methods rely mostly on shotgun-based approaches as they are easy to automate, allowing higher sample-throughput. However, stable-isotope labelling methods like iTRAQ™ or TMT™ are often too costly to be used for the sample throughput to cope with. On the other hand, SRM-based methods are restricted to a few dozen peptides / proteins, so that samples may have to be measured multiple times on the LC-MS-instrument to cover all potential marker proteins. In the future, the recently introduced label-free quantification methods like data-independent acquisition, SWATH or MSE may turn out to be best suited for this stage in biomarker research.
Proteomics for monitoring biomarkers
Mass spectrometry as an alternative to ELISA-based methods for the detection and quantification of known biomarkers has been established in recent years. While more expensive intially, biomarker monitoring by methods like scheduled selected reaction monitoring (sSRM) are not only more versatile and suited for the analysis of different biomarkers for distinct diseases24, but bear also the possibility of parallel detection of up to ~100 different proteins and therefore an entire ‘set of biomarkers’ that may only be conclusive in combination. The LC-MS-based methods like sSRMs will thus become an invaluable tool for diagnostics of diseases with a comprehensive protein regulation pattern in the future
Glossary of proteomics methods and approaches
Top-Down-Proteomics: Proteome analysis based on the separation of intact proteins3, frequently used methods are gel-based techniques as electrophoresis often grants higher resolution power than chromatographic methods on the proteome level. While top-down mass spectrometry (MS and MS/MS of intact proteins) can also be applied in these approaches which facilitate analysis of the overall modification state of a protein, they are not well suited for the analysis of highly complex mixtures. Therefore, most gel-based separation techniques are combined with subsequent bottom-up MS-techniques.
Bottom-Up-Proteomics: Proteome analysis based on separation and mass spectrometrical detection of peptides from proteolytic digests of entire proteomes25. The problem of separation is transferred from the protein level (like in top-down approaches) to the peptide level using mostly chromatographic separation methods. These approaches do not suffer from the biases typically hampering top-down gel-based analyses (see below); however, this advantage is countervailed by the inability to distinguish different protein isoforms.
In comparison to top-down MS-approaches, bottom-up mass spectrometry is particularly suited for analysis of proteins from high complexity mixtures. However, full sequence coverage of the identified proteins is typically not accomplished; thus, no conclusions can be drawn concerning the overall modification state of the proteins as particularly substoichiometric modifications may be overlooked.
Gel-based techniques: Electrophoretic techniques offer superb separation power in the analysis of protein mixtures. Gel-based methods have particularly gained wide popularity because they are versatile, easily implemented and highly reproducible. Different kinds of separations can be facilitated by this technique.
Separation of native proteins: Separation of proteins in the native state bears several advantages. The analysis of intact protein complexes by Blue-native or Colourless-native electrophoresis is a particular strength of this type of electrophoresis26. Furthermore, protein activities and protein folding are retained and can therefore be studied using this system. Moreover, the separations can be used as first dimension separations in multidimensional electrophoresis (see below).
Detergent-based separations: Besides the well-known and ubiquitously applied SDS-PAGE, further detergent-based systems can be used. In particular, cetyltrimethyl-ammonium-bromide (CTAB) and benzyl-dimethyl-n-hexadecyl-ammoniumchloride (16-BAC) as positively charged detergents have gained some popularity. Multidimensional separations based on two (or more) detergent-based separations are often used for the analysis of membrane proteins27.
Multidimensional, gel-based separations: The most widely applied two-dimensional electrophoresis is the combination of isoelectric focusing in the first and SDS-PAGE in the second dimension, commonly referred to as classical 2D-PAGE28. While it has the highest separation power and enables the separation and analysis of protein isoforms, it also has some drawbacks as it is biased against hydrophobic (membrane) proteins, strongly basic and high molecular weight proteins as well as low-abundant proteins. Therefore, different systems using detergents in both separation dimensions (such as CTAB/SDS-PAGE, 16-BAC/SDS-PAGE or 2D-SDS-PAGE) have been established for the analysis of membrane proteins. Furthermore, combinations of native and detergent-based separation dimensions have successfully been applied for the analysis of protein complex compositions.
Gel-free separation techniques: While gel-free separation systems for protein have been established, mostly so-called ‘shotgun’ or bottom-up –approaches account for the gel-free techniques29,30. The systems are typically multidimensional, HPLC-based separation systems such as IEX-RP or HILIC-RP. The reversed-phase chromatography is often used in the second dimension because of its superb compatibility to subsequent online-mass spectrometrical detection. While lacking the power to separate protein isoforms, it is compatible with a wide range of samples and particularly suited for the analysis of low-abundance protein from complex mixtures.
ESI-mass spectrometry techniques: Detection, identification and quantification of proteins and peptides by electrospray-ionisation-mass spectrometry is of central importance to proteomics analyses. Various different scan modi of the mass spectrometers facilitate a multitude of applications for different biological questions.
Information-/data-dependent acquisition (IDA/DDA): IDA or DDA methods are most often applied for the analysis of complex peptide mixtures in combination with up-front reversed phase HPLC. Thereby, a ‘full scan’ is recorded to detect peptides (‘precursor ions’) eluting from the HPLC. The precursor ions are subsequently isolated one at a time and fragmented (typically by ‘collisionally-induced dissociation’ (CID) or ‘electron-transfer dissociation’ (ETD)) and fragment ion spectra (also called MS/MS- or tandem mass spectra) are recorded31. After completion of such a ‘duty cycle’, the whole process is repeated starting with a further full scan. The MS/MS-spectra (together with the precursor ion masses from the full scan) are used afterwards for database searching, leading to protein identifications. Furthermore, quantitative data can be obtained from such analyses both using stable-isotope labelling techniques or label-free methods (see below).
Stable-isotope labelling: Various methods have been established using the exchange of naturally occurring isotopes like carbon-12 (12C) and nitrogen-14 (14N) with heavier stable isotopes (13C and 15N, respectively), also deuterated compounds are quite frequently used32. These heavy isotopes are introduced either by metabolic incorporation of heavy isotope-labelled amino acids or by chemical modification of the peptides by heavy isotope containing reagents. Since the chemical properties of the respective peptides / proteins do not change by the incorporation of heavy isotopes in comparison to the light counterparts, two different samples (one ‘heavy’ and one ‘light’ sample) can be mixed up-front and processed together throughout all separation processes, thereby assuring the exactly identical sample treatment of both samples during the analysis. Both samples can be distinguished and differentially quantified using mass spectrometry in the end. Usage of different combinations of stable isotopes enable also the parallel processing of more than two samples, called ‘multiplexing’ of samples.
Labelling-free approaches: Since stable-isotope labelling of samples requires additional sample treatment and is typically quite costly, label-free approaches have been established33. Particularly, the below-mentioned mass spectrometric methods are capable of highly sensitive, high accuracy detection and quantification of peptides from complex samples.
Selected reaction monitoring, also called multiple reaction monitoring34, is a highly sensitive method for quantification of known peptides by specific isolation of both the precursor ion (the intact peptide) and specific fragment ions. The sensitivity and good quantification accuracy of the method is countervailed by the fact that peptides of interest have to be known prior to the analysis and only limited numbers of peptides per run can be quantified. So-called ‘scheduled’ SRM-methods use also the known HPLC-retention times of the peptides to enhance the number of quantifiable peptides to more than 100.
Hyper-reaction monitoring (SWATH), Data-independent acquisition (DIA) and MSE: All these methods allow label-free quantification of peptides from highly complex mixtures35. In contrast to SRM-methods, not individual precursor ions but broader mass windows (from a few to several hundred Dalton) are isolated and fragmented together. Typically, more than one peptide is fragmented in parallel and high mass resolution is needed to be able to specifically quantify the fragment ions without interference from further fragment ions generated from precursors that were fragmented in parallel. While this technique is not as sensitive and makes much higher demands on mass spectrometry equipment (typically QTOF- or orbitrap-mass spectrometers) in comparison to SRM methods (typically accomplished on triple-quadrupole mass spectrometers), it enables the analysis of up to several thousand peptides in one run, also making these techniques particularly versatile in biomarker studies.
References
- Rehm, H., Proteinbiochemie/Proteomics, Spektrum Akademischer Verlag, Heidelberg 2000, pp. 22-23
- Yates, J. R., 3rd, Kelleher, N. L., Anal Chem 2013, 85, 6151
- Zhou, H., Ning, Z., Starr, A. E., Abu-Farha, M., Figeys, D., Anal Chem 2012, 84, 720-734
- Shevchenko, A., Zachariae, W., Biochem Soc Trans 1999, 27, 549-554
- Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., Yang, L., Wolting, C., Donaldson, I., Schandorff, S., Shewnarane, J., Vo, M., Taggart, J., Goudreault, M., Muskat, B., Alfarano, C., Dewar, D., Lin, Z., Michalickova, K., Willems, A. R., Sassi, H., Nielsen, P. A., Rasmussen, K. J., Andersen, J. R., Johansen, L. E., Hansen, L. H., Jespersen, H., Podtelejnikov, A., Nielsen, E., Crawford, J., Poulsen, V., Sorensen, B. D., Matthiesen, J., Hendrickson, R. C., Gleeson, F., Pawson, T., Moran, M. F., Durocher, D., Mann, M., Hogue, C. W., Figeys, D., Tyers, M., Nature 2002, 415, 180-183
- Tulp, A., Verwoerd, D., Neefjes, J., J Chromatogr B Biomed Sci Appl 1999, 722, 141-151
- Schagger, H., von Jagow, G., Anal Biochem 1991, 199, 223-231
- Schagger, H., Cramer, W. A., von Jagow, G., Anal Biochem 1994, 217, 220-230
- Reisinger, V., Eichacker, L. A., Methods Mol Biol 2012, 854, 343-353
- Peters, K., Braun, H. P., Methods Mol Biol 2012, 854, 145-154
- Bich, C., Zenobi, R., Curr Opin Struct Biol 2009, 19, 632-639
- Tallet, B., Astier-Gin, T., Castroviejo, M., Santarelli, X., J Chromatogr B Biomed Sci Appl 2001, 753, 17-22
- Stahl, S., Reinders, Y., Asan, E., Mothes, W., Conzelmann, E., Sickmann, A., Felbor, U., Biochim Biophys Acta 2007, 1774, 1237-1246
- Reinders, J., Sickmann, A., Proteomics 2005
- Mann, M., Jensen, O. N., Nature Biotechnology 2003, 21, 255 – 261
- Gibson, B. W., Int J Biochem Cell Biol 2005, 37, 927-934
- Reinders, J., Sickmann, A., Biomol Eng 2007, 24, 169-177
- Ivakhno, S., Kornelyuk, A., Biochemistry (Mosc) 2006, 71, 1060-1072
- Bolwell, G. P., Slabas, A. R., Whitelgge, J. P., Phytochemistry 2004, 65, 1665-1669
- Calligaris, D., Villard, C., Lafitte, D., J Proteomics 2011, 74, 920-934
- Gramolini, A. O., Peterman, S. M., Kislinger, T., Clin Pharmacol Ther 2008, 83, 758-760
- Cowan, M. L., Vera, J., Expert Rev Proteomics 2008, 5, 21-23
- Craft, G. E., Chen, A., Nairn, A. C., Methods 2013, 61, 186-218
- Calvo, E., Camafeita, E., Fernandez-Gutierrez, B., Lopez, J. A., Expert Rev Proteomics 2011, 8, 165-173
- Han, X., Aslanian, A., Yates, J. R., 3rd, Curr Opin Chem Biol 2008, 12, 483-490
- Krause, F., Seelert, H., Curr Protoc Protein Sci 2008, Chapter 14, Unit 14 11
- Braun, R. J., Kinkl, N., Beer, M., Ueffing, M., Anal Bioanal Chem 2007, 389, 1033-1045
- Gorg, A., Weiss, W., Dunn, M. J., Proteomics 2004, 4, 3665-3685
- Abdallah, C., Dumas-Gaudot, E., Renaut, J., Sergeant, K., Int J Plant Genomics 2012, 2012, 494572
- Dumont, D., Noben, J. P., Verhaert, P., Stinissen, P., Robben, J., Proteomics 2006, 6, 4967-4977
- Steen, H., Mann, M., Nat Rev Mol Cell Biol 2004, 5, 699-711
- Putz, S., Reinders, J., Reinders, Y., Sickmann, A., Expert Rev Proteomics 2005, 2, 381-392
- Wong, J. W., Cagney, G., Methods Mol Biol 2010, 604, 273-283
- Kiyonami, R., Domon, B., Methods Mol Biol 2010, 658, 155-166
- Hopfgartner, G., Tonoli, D., Varesio, E., Anal Bioanal Chem 2012, 402, 2587-2596
Biography
 Joerg Reinders studied biochemistry at the Ruhr-University Bochum, and completed his diploma thesis at the Medical Proteome Center on the analysis of protein phosphorylations. He obtained his PhD at the Julius-Maximilians-University Wuerzburg in the field of yeast mitochondrial proteomics. Changing the model system of interest, he worked on proteome analysis of plants and insect before becoming group leader for proteomics at the Institute of Functional Genomics at the University Regensburg. His current main topic is elucidation of molecular (patho-) mechanisms underlying cellular processes and diseases using proteomics as a versatile tool in the analysis of the cellular network.
Joerg Reinders studied biochemistry at the Ruhr-University Bochum, and completed his diploma thesis at the Medical Proteome Center on the analysis of protein phosphorylations. He obtained his PhD at the Julius-Maximilians-University Wuerzburg in the field of yeast mitochondrial proteomics. Changing the model system of interest, he worked on proteome analysis of plants and insect before becoming group leader for proteomics at the Institute of Functional Genomics at the University Regensburg. His current main topic is elucidation of molecular (patho-) mechanisms underlying cellular processes and diseases using proteomics as a versatile tool in the analysis of the cellular network.
Issue
Related topics
Assays, Biomarkers, HTS (High Throughput Screening), Liquid Chromatography - Mass Spectrometry (LC-MS), Proteomics
