

Cellular senescence as an anti-tumour mechanism
Posted: 24 June 2010 |
One of the critical steps in human carcinogenesis is cellular immortalisation, a process in which cells must escape senescence and acquire an infinite lifespan. In the absence of immortalisation, although a cell might undergo malignant transformation, it could not proliferate indefinitely. Furthermore, it has been clearly established in vitro and in vivo that cellular senescence is a tumour suppressor mechanism induced by oncogenic stress. Normal somatic cells grown in culture cease to proliferate, senesce, after a finite number of divisions.
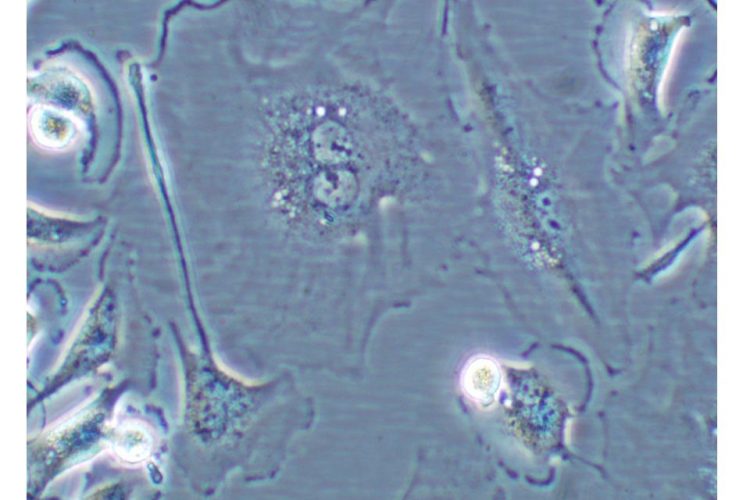
Figure 1 Cell undergoing senescence. Cell showing characteristics of cellular senescence: binucleated and flat and enlarged morphology
One of the critical steps in human carcinogenesis is cellular immortalisation, a process in which cells must escape senescence and acquire an infinite lifespan. In the absence of immortalisation, although a cell might undergo malignant transformation, it could not proliferate indefinitely. Furthermore, it has been clearly established in vitro and in vivo that cellular senescence is a tumour suppressor mechanism induced by oncogenic stress1. Normal somatic cells grown in culture cease to proliferate, senesce, after a finite number of divisions.
This phenomenon, first described by Hayflick in 19652, is referred to as replicative senescence3. Senescent cells generally have a large, flattened morphology (see Figure 1).
These cells are growth arrested in G1 phase of the cell cycle, are incapable of synthesising DNA and are unresponsive to stimulation with growth factors; yet interestingly, are still metabolically active. Senescent cells can be distinguished from pre-senescent, immortal, quiescent or terminally differentiated cells by histochemical detection of the biomarker -galactosidase (SA -gal) at pH 6. Other markers of cellular senescence include p16INK4a, p21CIP1/WAF1, PAI-1, phosphorylated H2AX, activated CHK1 and CHK2.
In normal human cells, telomeres become progressively shorter with each population doubling until they reach a critically short length. The progressive shortening of telomeres in normal somatic cells is more prominent than in germ cells, and to a lesser extent stem cells, where the telomeres are maintained by telomerase4. Critically, short telomeres may be one of the signals that can induce senescence. Inactivation of p53, pRB/p16INK4a or another key proliferative checkpoint gene can extend the lifespan of a cell beyond senescence. Ectopic expression of the catalytic unit of telomerase, hTERT, can stabilise telomere length allowing cells to grow indefinitely5. In addition to telomere dysfunction, cellular senescence can also be elicited by other types of stress, including oncogene activation6. This phenomenon is not observed for oncogenic RAS exclusively; many – but not all – of its effectors, including activated mutants of RAF, MEK and BRAF, were shown to cause senescence as well. Some oncogenes, like RAS, CDC6, cyclin E and STAT5, which induce senescence, also trigger a DNA-damage response, which is associated with DNA hyper-replication and appears to be causally involved in oncogene-induced senescence in vitro.
During most of the last decade, oncogene-induced senescence has been studied predominantly in cell culture systems, triggering a long debate as to whether or not oncogene-induced senescence corresponds to a physiologically relevant phenomenon in vivo. In favour of oncogene-induced senescence representing an in vitro phenomenon only is that artificial conditions, such as the use of bovine serum and plastic dishes, as well as the presence of supraphysiologic O2, generate a stress signal that at the very least contributes to triggering a cellular senescence response. However, on the contrary, senescence bypass screens have identified several genuine human oncogenes including TBX2, BCL6, KLF4, hDRIL, BRF1, PPP1CA, SAHH and others. Furthermore, virtually all human cancers lack functional p53/pRB pathways, two key senescencesignalling routes7. In addition, human cancers often carry mutations in sets of genes, which are known to collaborate in vitro in bypassing the senescence response. In 2005, four groups simultaneously reported the protective physiological role of oncogene-induced senescence in vivo. This was shown for murine lung adenomas, T-cell lymphomas, prostate tumours, as well as human benign melanocytic nevi (‘moles’)8-10. In a mouse model, RASV12 knockin mice were shown to develop lung adenomas that were characterised by a low proliferative index and elevation of SA-β-Gal activity and other senescence markers10. The few adenocarcinomas that did emerge showed considerable proliferative activity and were lacking senescence markers.
Schmitt and colleagues characterised RASV12-driven mouse T-cell lymphomas in which apoptosis was blocked; these lesions entered senescence after drug therapy, which was dependent on the chromatin-remodeling enzyme Suv39h18. On the other hand, nevi are benign melanocytic tumours that generally lack proliferative activity. They commonly suffer from an activating mutation in the BRAF protein kinase, with a single amino acid change (V600E) accounting for most of the cases. Few nevi grow larger than one centimetre, and less than one per 1000 ever progresses to melanoma, the highly malignant counterpart of a benign nevus. Michaloglou and collaborators showed that the cell cycle arrest of human nevi has the hallmarks of oncogeneinduced senescence: nevi undergo long-term cell cycle arrest, carry an activated oncogene product (BRAFE600), express elevated levels of p16INK4a and display increased SA-β-Gal activity. In addition, they do not show detectable signs of telomere erosion, suggesting that they have undergone oncogene-induced senescence9.
Extrinsic factors such as anticancer agents, γ-irradiation or UV light have been shown to induce premature senescence as a DNA damage–mediated cellular stress response.

Figure 1 Cell undergoing senescence. Cell showing characteristics of cellular senescence: binucleated and flat and enlarged morphology
Senescence genes and pathways
Cellular senescence pathways are believed to have multiple layers of regulation with additional redundancy built into these layers. Many of the functional studies, where a putative senescence gene is over expressed in cells, indicate that although multiple genes/pathways may be abrogated in a particular cell line, as little as one gene/pathway is required for repair and subsequent reversion to senescence.
Pathways known to regulate cellular senescence/immortalisation, including the p16INK4a/pRB pathway, the p19ARF/p53/ p21CIP1/WAF1 pathway and the PTEN/p27KIP1 pathway are reviewed11. Other genes that have been shown to induce a senescence-like phenotype include PPP1A, SAHH, Csn2, Arase and BRF1, PGM, IGFBP3 and IGFBPrP1, PAI-1, MKK3, MKK6, Smurf2 and HIC-5. All these genes have shown to be related to human tumorigenesis. However, all these genes and pathways, as we indicated earlier, can act in sequential steps establishing a well regulated process.
Over all steps, DNA methylation regulates the expression of senescence genes, with the capability of controlling the process. In human cancers, the silencing of tumour suppressor genes through aberrant DNA methylation of a CpG island(s) in the promoters in these genes is a common epigenetic change. There are an assortment of pathways from which genes have been shown to be hypermethylated in cancer cells, including DNA repair, cell cycle control, invasion and metastasis.
Senescence based therapy
Clearly, from a therapeutic perspective senescence restoration would represent an interesting option, just as the ability of p53 to induce apoptosis in established tumours is believed to be essential for cancer therapy12. Two groups recently developed elegant mouse models to study the consequences on cellular senescence of the restoration of p53 function on tumorigenesis. In a model for p53-dependent liver cancer, Lowe and collaborators studied the effect of p53 restoration in established liver carcinomas13. Tumorigenic hepatoblasts expressing a conditional p53 short interfering RNA produced invasive hepatocarcinomas, but soon after re-expression of p53 the tumours underwent dramatic regression. This was irreversible and, remarkably, did not depend on p53-induced apoptosis. Instead, restoring endogenous p53 function triggered cellular senescence. The senescent cells acquired a specific gene expression profile signature that included the upregulation of inflammatory cytokines. It was shown that this led to the activation of the innate immune system, clearing the tumour, thereby establishing a link between the cellular senescence program and the innate immune system in suppressing tumorigenesis. Similarly, Ventura et al. generated a re-activatable p53-deficient mouse model for lymphomas and osteosarcomas. Both tumour types regressed completely after p53 restoration14. Interestingly, whereas re-expression of endogenous p53-triggered apoptosis in lymphomas, it led to a senescencelike cell cycle arrest in the osteosarcomas, pointing to an important role for tissue type and/or genetic context in determining the cellular response in tumour regression.
The treatment of different tumour cell lines with different chemotherapeutic agents, radiation or differentiating agents induces irreversible growth arrest, with enzymatic and morphologic changes resembling those occurring during replicative senescence. Treatment of normal human foreskin and lung fibroblasts with DNA-damaging drugs such as bleomycin or actinomycin D induced an irreversible cell-cycle arrest with a senescencelike phenotype including a transient upregulation of p53 and p21 protein levels, followed by increased p16INK4a protein expression and detectable SA-β-Gal activity. Similarly, exposure of adenocarcinoma cells to topoisomerase inhibitors produced premature senescence that was initially accompanied by overexpression of p53 and p21, whereby the subsequent overexpression of p16INK4a persisted after drug withdrawal, highlighting the role of p16INK4a in maintenance of the growth arrest. Different chemical agents can induce cellular senescence epigenetically. Treatment of primary cells with H2O2 or butyrate provokes early senescence. Similar results were obtained after treatment with high doses of radiation and other damaging agents. On the other hand, moderate doses of doxorubicine induced a senescent phenotype in 11 out of 14 tumour cell lines analysed, independently of p53 status. A similar effect has been observed in lines from human tumours treated with cisplatin, hydroxyurea and bromodeoxyuridine. In mammary carcinoma cell lines treated in vitro and in vivo with differentiating agents, terminal proliferative arrest with minimal toxicity for normal cells has been observed. The propensity of tumour cells to undergo senescence in response to different kinds of damage induced by commonly used chemotherapeutic treatments was compared on cell lines from different tumour origins. Under equitoxic doses, the strongest induction of a senescent phenotype was observed with DNA-interacting agents (doxorubicin, aphidicolin and cisplatin) and the weakest effect was observed with microtubule-targeting drugs (Taxol and vincristine). A medium response was observed with ionising radiation, cytarabine and etoposide. Induction of senescence by the drugs was dose dependent and correlated with the growth arrest observed in the cultures. The drug-induced senescent phenotype in tumour cells was not associated with telomere shortening and was not prevented by the expression of telomerase.
Drug-induced senescent phenotypes have been confirmed in vivo. A study from Poele et al.15 revealed the correlation between chemotherapeutic treatment in clinical cancer and the senescence response. In frozen samples from breast tumours treated by neo – adjuvant chemotherapy (cyclophosphamide, doxorubicin and 5-fluoracyl), senescent markers were detected in 41 per cent of samples from treated tumours. Normal tissue was negative, suggesting that the chemotherapy-induced senescence was a specific response of tumour cells. Interestingly, senescence response was associated with wild type p53 and the increased expression of p16.
Cellular senescence retains the potential to be used as a drug-effector program. However, it is a complex biological phenomenon with the need for further investigations.
References
- Campisi J (2001). Cellular senescence as a tumour-suppressor mechanism. Trends Cell Biol 11, S27-31
- Hayflick L (1965). The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 37, 614-636
- Coates PJ (2002). Markers of senescence? J Pathol 196, 371-373
- Cong YS, Wright WE, Shay JW (2002). Human telomerase and its regulation. Microbiol Mol Biol Rev 66, 407-425, table of contents
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349-352
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593-602
- Malumbres M, Carnero A (2003). Cell cycle deregulation: a common motif in cancer. Prog Cell Cycle Res 5, 5-18
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA (2005). Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436, 660-665
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720-724
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP (2005). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumourigenesis. Nature 436, 725-730
- Berube NG, Smith JR, Pereira-Smith OM (1998). The genetics of cellular senescence. Am J Hum Genet 62, 1015-1019
- Evan GI, Christophorou M, Lawlor EA, Ringshausen I, Prescott J, Dansen T, Finch A, Martins C, Murphy D (2005). Oncogene-dependent tumour suppression: using the dark side of the force for cancer therapy. Cold Spring Harb Symp Quant Biol 70, 263-273
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656-660
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661-665
- Poole JC, Thain A, Perkins ND, Roninson IB (2004). Induction of transcription by p21Waf1/Cip1/Sdi1: role of NFkappaB and effect of non-steroidal anti-inflammatory drugs. Cell Cycle 3, 931-940
About the Author
Amancio Carnero has a PhD in Molecular Biology from CSIC, whose laboratory actively works in translational research in cancer. During his career, Carnero worked at Cold Spring Harbor Laboratory (USA), University College London (UK) and Spanish National Cancer Research Centre (CNIO, Madrid). His work always follows a single line: to understand the molecular mechanisms of cancer and trying to apply this knowledge to discover new anti-tumour therapies. Currently, his research at IBIS (Seville) focuses in the identification and characterisation of genes with diagnostic or therapeutic relevance in cancer, the establishment of causality in the tumoural process and the validation of new therapeutic targets. Contact the author: [email protected]
