

The evolution of RNAi technologies in the drug discovery business
Posted: 29 October 2010 |
In the past decade, the pharmaceutical industry has exploited the naturally occurring cellular RNAi pathway to enhance drug discovery research. The RNAi pathway, triggered by dsRNA, selectively, although not always specifically, degrades mRNA leading to substantial decreases in post-transcriptional gene expression1. Researchers have capitalised on this intrinsic pathway by synthesising RNAi reagents to modify the expression of any desired gene. RNAi libraries consisting of synthetic siRNAs or plasmid based shRNAs are amendable to largescale genome-wide screening campaigns to search for new therapeutic targets. Such loss of function screens can reveal novel targets and synthetic lethal interactions for cancer therapy2,3. These screens have also been used to identify novel host factors for diseases such as Hepatitis C4-7 and HIV8-14. Selective gene silencing can deconvolute molecular pathways implicated in disease onset and progression15.
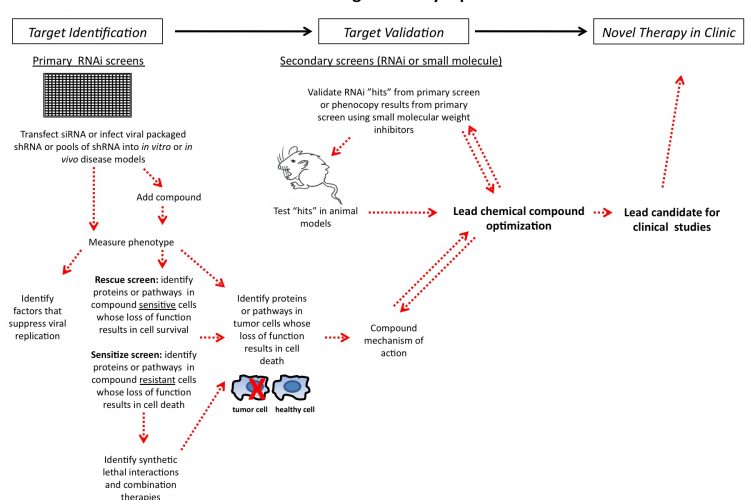
Figure 1 siRNA drug discovery pipeline
In the past decade, the pharmaceutical industry has exploited the naturally occurring cellular RNAi pathway to enhance drug discovery research. The RNAi pathway, triggered by dsRNA, selectively, although not always specifically, degrades mRNA leading to substantial decreases in post-transcriptional gene expression1. Researchers have capitalised on this intrinsic pathway by synthesising RNAi reagents to modify the expression of any desired gene. RNAi libraries consisting of synthetic siRNAs or plasmid based shRNAs are amendable to largescale genome-wide screening campaigns to search for new therapeutic targets. Such loss of function screens can reveal novel targets and synthetic lethal interactions for cancer therapy2,3. These screens have also been used to identify novel host factors for diseases such as Hepatitis C4-7 and HIV8-14. Selective gene silencing can deconvolute molecular pathways implicated in disease onset and progression15.
The use of RNAi screening is an efficient and cost effective method for identifying possible targets for low molecular weight compounds or antibody therapies. RNAi has been demonstrated as a powerful tool for both the identification and validation of genes in cell based assays and in in vivo applications16. Recent developments in RNAi formulation and delivery17,18 have advanced the use of RNAi as a therapeutic for targets that would otherwise not be druggable using conventional methods. This review discusses how RNAi has contributed to the drug discovery process and the future possibilities of using RNAi in drug discovery and as a therapeutic modality itself.
RNAi is the mechanism by which dsRNA inhibits gene expression. A dsRNA molecule is processed by the enzyme DICER into a small 19 – 23 base pair duplex known as a shortinterfering RNA (siRNA). The siRNA then enters a multi-protein complex known as the RNA induced silencing complex (RISC). Once incorporated into RISC, a helicase unwinds the sense and antisense strands of the siRNA. The antisense strand then binds the complementary mRNA. The target mRNA is cleaved by an RNase inside the RISC complex, which results in silenced gene expression through increased mRNA degradation and reduced protein production. Researchers can utilise chemically synthesised siRNAs that are designed to target a specific gene of interest. The creation of genome-scale siRNA libraries allows for the potential identification of new drug targets that regulate disease relevant pathways.
Traditionally, siRNAs are transfected into target cells lines via a reagent that carries the siRNA across the lipid bilayer of the cell. The transfection conditions need to be optimised in order to ensure the phenotype being screened is appropriately measured and that the signal-to-background ratio is above a certain quantifiable threshold. There are numerous limitations preventing the accurate measurement of mRNA levels used to assess small interfering RNA (siRNA) efficacy after transfection. One approach to overcome these difficulties is to use a phenotypically neutral assay to quantify the knockdown of an exogenous luciferase gene delivered by a lentiviral vector19. In this assay, the efficacy of a luciferase siRNA is compared to a negative control siRNA across many distinct assay parameters including cell type, cell number, lipid type, lipid volume, time of the assay and concentration of siRNA. Once the siRNA transfection is optimised as a 384-well luciferase knockdown, the biologically relevant phenotypic analysis can proceed using the optimal siRNA transfection conditions. While this and other similar approaches20 play a crucial role in RNAi screening assay development, certain cell lines such as primary cells are not easily transfectable using lipid complex mediated uptake. One way researchers overcome this hurdle is to use RNAi libraries comprised of vector based plasmids encoding short-hairpin RNA (shRNA). Either constitutive or inducible shRNA hairpin vectors can be integrated stably into the genome for long term or regulatable knock down studies.
Both formats of RNAi libraries, either siRNA or shRNA, have their advantages and disadvantages. SiRNAs are straightforward to design and synthesise at scale for screens. The siRNA reagents are most useful in transfectable cell lines but gene silencing is transient. SiRNAs can also give rise to off target effects which can lead to undesired phenotypes making it difficult to interpret the results21. In contrast, shRNAs in viral vectors have the advantage of being transduced into difficult to transfect cell lines. The hairpin can be stably integrated into the host genome resulting in long term gene silencing. This makes them more suitable for assays that require longer incubation times such as soft agar, colony formation or in vivo xenograft studies. They can be cloned into inducible vectors, allowing for regulated expression of the respective target gene22. A major disadvantage of viral vectors is the requirement of a viral packaging system and special bio-safety facilities required for amplification. ShRNA libraries are amendable to pooled screening strategies where multiple shRNAs are pooled and assayed in parallel in contrast to running each shRNA in an individual well9,23-27. A well-by-well based shRNA screen is greatly limited by the upfront efforts to design and validate multiple shRNAs targeting each gene separately.

Figure 1 siRNA drug discovery pipeline
Another application of RNAi screening is performing screens in a format more compatible with in vivo growth conditions. Demonstration of anchorage independent growth of transformed cancer cells grown in a semi-solid, 3-dimensional (3D) medium, such as soft agar, is an ideal assay for determining tumorigenicity of cells in vitro. The soft agar assay (SAA) can yield results that closely match those of xenograft assays where tumorigenic cells are injected into nude mice28. This is done to discover genes that regulate anchorage-independent growth in cancer cells that traditional 2D growth assays might miss. Performing SAAs in a high throughput manner has been shown to be both tech nically feasible and robust29,30. These protocols are currently being used to screen cell lines that are sensitive to anti-cancer compounds when grown in a 3D format. Running soft agar assays in high throughput RNAi screens is a more physiologically relevant way to identify pathways and proteins whose decreased expression leads to slowed or inhibited tumour growth than traditional in vitro based assays run on plastic.
The miniaturisation of cell based assays to 1536 well format has greatly increased the throughput capacity cell based RNAi assay31. The smaller scale format uses fewer reagents thereby significantly reducing screening time and costs32. To date, successful high density chemical compound screens have been successfully run in 1536 well format where greater than one million small molecular compounds were analysed in one screen33. The use of high density, ultra high-throughput RNAi screens in this format opens the possibilities for expanded coverage of siRNAs used and conditions tested. The increase in the number of wells that can be screened at once leads to the novel screening method of matrixed based siRNA screening where combinations of genes whose loss of function reveal desired phenotypes. Continued advancements in automated microscopy platforms and the use of fluorescence tags and antibodies have allowed researchers to perform higher throughput high content RNAi screens to identify pathways and proteins that modify cellular function. Advanced time-resolved live cell imaging assays can advance the understanding of complex cellular processes that cannot be studied using traditional endpoint assays. Researchers can now perform genome-wide RNAi screening assays that are informative on cell growth rates, migration, subcellular localisation, proteinprotein interactions and when monitoring dynamic changes in intracellular structures34.
An example of where RNAi screening has been successful at identifying drug targets is in the area of host factors that regulate viral replication. Novel host factors that regulate viral replication is a new paradigm to treat multiple genotypes of a pathogenic virus and a strategy to overcome viral resistance induced by therapeutics directed to viral proteins. Several RNAi based screens have revealed an important role of phosphatidylinositol-4-kinase III alpha (PI4KCA) in HCV replication5-7. Although the RNAi libraries used by each research group varied in design and format, PIK4CA was consistently identified as a host factor that potently regulated HCV replication. Pharmacological inhibition of this enzyme may represent a new class of antiviral therapy for multiple genotypes of HCV. Other siRNA and shRNA screens have also identified other cellular host factors critical for all stages of the HIV life cycle8,9,11-14. These studies have yielded a significant number of known and novel host factors important for HIV replication that could be further developed as therapeutic targets. Thus, RNAi based screening has led to new insights into the complexity of HCV and HIV life cycles and the host factors that regulate them. The new pathway insights identified through these screens increases the potential for novel antiviral strategies to be explored10.
RNAi screening has been an essential tool for discovering genes and / or pathways that drive cancer cell survival. Selective silencing of survival pathways can mimic the effect of a targeted cancer therapy leading to selective killing of cancer cells that harbour certain mutations. Loss of function screens combined with a cell viability endpoint assay plays a significant role in the identification of novel tumour growth factor pathways35. RNAi screens have also been used in combination with compound treatment to identify the mechanism of action of compounds in late stage drug development pipelines. Successful target gene silencing should confer resistance or be ‘rescued’ from compound induced cell death. For example, RNAi based screens have uncovered the mechanism of action of SMAC mimetic compounds36-38. These studies have demonstrated that TNFα and the NKκB pathway are critical to the activity target for SMAC mimetic compounds. Conversely, compound sensitisation RNAi screens are used to identify target genes that, when knocked down, can increase the efficacy of the compound in resistant cell lines39. This is particularly important for targeted therapies where understanding the MOA is critical.
RNAi screening can also identify genes that, when inhibited, result in greater sensitivity to current cancer standard of care chemotherapy. A partial response or resistance to chemo – therapies is a major hurdle in the successful treatment of cancers. Improvements to existing therapies through new drug combinations are of high clinical interest. Paclitaxel has been used extensively for treating multiple forms of cancers; however, many patients develop resistance to treatment. Recent RNAi screens have identified genes whose loss of function enhances sensitivity to Paclitaxel in breast cancer cells40. Specific chemical inhibitors against the top proteins identified in these screens showed synergistic drug activity when used in combination with Paclitaxel3. Lower doses of chemotherapies can be used in combination with specific RNAi silencing of genes known to be highly over-expressed in tumour cells. George et al showed that knockdown of the BCL-2 gene sensitises human glioblastoma cells to Paclitaxel treatment and could present a novel approach to treatment of malignant brain tumours41. Irons et al showed the utility in performing parallel compound and RNAi screens in combination with Tamoxifen treatment, an inhibitor of the oestrogen receptor used to treat breast cancer. These high throughput screening efforts led to the discovery that targeting of the PDK-1 signalling pathway via siRNA or with small molecular inhibitors increased sensitivity to Tamoxifen42.
A synthetic lethal interaction is when the combined inhibition or mutation of two or more genes results in cell death whereas cells survive perturbation of either gene alone. RNAi screening is well suited to identify these interactions. For example, several groups are using pooled shRNA strategies to identify proteins that are synthetic lethal in both cell and in vivo based experiments. Large pools of shRNAs are introduced into target cells lines over time and the cells that survive are deep sequenced to ‘retrieve’ shRNAs present in the surviving cells. Another technique is to measure shRNA enrichment using a barcode microarray where specific DNA sequences are barcodes in the shRNAs23-25. Several research groups have used this approach to identify synthetic lethal interactions with the KRAS oncogene26,43,44. Additionally, shRNA pooling strategies have also been utilised for in vivo to explore tumour growth in mouse models of cancer16. In this study, researchers were able to screen nearly 1,000 genetics interrogations in one tumour bearing mouse.
SiRNAs have also proven to be efficacious in mouse models. Huang and colleagues were able to develop an intraperitoneal lipidoid delivery system to target Claudin-3 (CLDN3) siRNAs to ovarian xenografts tumour cells in mice45. CLDN3 has been shown to suppress tumour growth in ovarian cancer cells in vitro46. The successful silencing of CLDN3 in vivo resulted in reduced cell proliferation and tumour growth, an increase in apoptotic cells, and reduced metastasis. This suggests that lipidoid delivery of siRNAs in vivo has the potential to become a therapeutic in ovarian as well as other forms of cancer that specifically over express proteins that can be targeted by in vivo siRNAs. Localised or topical delivery of siRNAs, that is delivery of siRNAs directly to damaged tissues, has shown to be well suited for treating diseases associated with the eyes, skin or mucosal membranes as the delivery formulations continue to improve18. Systemic delivery of siRNAs pose some significant challenges as the siRNA therapeutics need to travel through the patient’s body and to the target organ(s), evading uptake by nontargeted tissues. Systemic delivery of siRNAs needs to be carried out in vehicles that remain stable in the blood and endothelial system17. siRNA therapies are moving into clinical trials to treat diseases such as age related macular degeneration, diabetic macular edema, respiratory syncytial virus (RSV), hepatitis B (HBV), HIV and therapeutic siRNAs targeting solid tumours47.
The complex regulatory network of microRNAs (miRNAs) presents a challenge to develop drugs to target these molecules, including the identification of novel tumour suppressors and the pathways they regulate. MicroRNAs (miRNAs) are small non-coding, single stranded RNAs that regulate gene expression post transcriptionally by binding near complementary protein mRNA sequences thereby inducing RISC mediated mRNA degradation. MiRNAs can also regulate gene expression by binding to imperfect complementary sequences in the 3’ UTR of the target mRNA which also triggers a decrease in gene expression through a similar RISC complex used in the RNAi pathway. Researchers are setting up platforms and using miRNAs as a way to explore biomarkers of disease and response to drug treatments48,49. MiRNA profiling of cancer cells and primary tumour samples have found miRNAs that regulate tumour progression and metastasis in breast cancer49-51. MiRNAs provide a new class of molecules to study in the hopes of identifying agents that play a role in development of human diseases and a new area for drug companies to explore for novel biomarkers and potential therapeutic opportunities.
In summary, the use of siRNA reagents has evolved from a research tool into a promising therapeutic. RNAi has the ability to be used as an effective screening tool to interrogate all the genes of the human genome to answer questions about the mechanism of action of compounds and disease processes. Identification of novel drug targets, the pathways that regulate compound resistance, synthetic lethal interactions and genes that are essential for disease progression are essential to the drug discovery pipelines and the betterment of human health. RNAi can be used to unravel the mechanism of action of compounds being developed in the drug discovery pipeline, helping pharmaceutical companies deliver more novel and higher quality therapies into the clinic. Focused research in the design and delivery of siRNA in vivo provides a promising new field of therapeutics that may be able to drug the undruggable targets.
References
1. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391: 806-811
2. Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, Erickson H, Kovacsovics T, O’Hare T, Heinrich MC, Druker BJ. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci USA 2009; 106: 8695-8700
3. Bauer, JA, Ye F, Marshall CB, Lehmann BD, Pendleton CS, Shyr Y, Artega CL, Pietenpol JA. RNA interference (RNAi) screening approach identifies agents that enhance paclitaxel activity in breast cancer cells. Breast Cancer Research 2010; 12:R41
4. Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton-Davies J, Wilson C, Myer V, CornellaTaracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither L. Class III Phosphatidylinositol 4-Kinase Alpha and Beta are novel host factor regulators of hepatitis C virus replication. J Virol 2009; 83: 10058-10074
5. Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, Cordingley MG, Kukolj G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009; 387: 5-10
6. Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA 2009; 106:7577-82
7. Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009; 5: 298-307
8. Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008; 319; 921-926
9. Rato S, Maia S, Brito PM, Resende L, Pereira CF, Moita C, Freitas RP, Moniz-Pereira J, Hacochen N, Moita LF, Goncalves J. Novel HIV-1 knockdown targets indentified by an enriched kinases/phosphatases shRNA library using a long-term iterative screen in Jurkat cells. PloS ONE 2010; 5: e9276
10. Pache L, König R, Chanda SK. Identifying HIV-1 host factors by genome-scale RNAi screening. Methods 2010 [Epub ahead of print]
11. König R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008; 135: 49-60
12. Yamagishi M, Ishida T, Miyake A, Cooper DA, Kelleher AD, Suzuki K, Watanabe T. Retroviral delivery of promoter-targeted shRNA induces long-term silencing of HIV-1 transcription. Microbes Infect 2009; 11: 500-508
13. Yeung ML, Houzet L, Yedavalli VS, Jeang KT. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem 2009; 284: 19463-19473
14. Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008; 4: 495-504
15. Smith JA, White EA, Sowa ME, Powell MLC, Ottinger M, Harper JW, Howley PM. Genome wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci USA 2010; 107: 3752-3757
16. Meacham CE, Ho, EE, Dubrovsky E, Gertler FB, Hemann MT. In vivo RNAi screening identifies regulators of actin dynamics as key determinants of lymphoma progression. Nature Genet 2009; 41: 1133-1137
17. Baigude H, Rana TM. Delivery of therapeutic RNAi by nanovehicles. Chem BioChem 2009; 10: 2449-245
18. Whitehead KA, Langer R, Anderson Dg. Knocking down barriers: advances in siRNA delivery. Nature Rev. Drug Discov. 2009; 8: 129-138
19. Borawski J, Lindeman A, Buxton F, Labow M, Gaither LA. Optimization procedure for small interfering RNA transfection in a 384-well format. Biomol Screen 2007; 12: 546-559
20. Carralot JP, Kim TK, Lenseigne B, Boese AS, Sommer P, Genovesio A, Brodin P. Automated high-throughput siRNA transfection in raw 264.7 macrophages: A case study for optimization procedure. J Biomol Screen 2009; 14: 151-160
21. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nature Rev Drug Discov 2010; 9: 57-67
22. Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao Y, Lengauer C, Sellers WR, Benson JD. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009; 8: 498-504
23. Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A lossof- function RNA interference screen for molecular targets in cancer. Nature 2006; 441: 106–110
24. Brummelkamp TR, Fabius AW, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol 2006; 2: 202-206
25. Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, Westbrook TF, Liang AC, Chang K, Hackett JA, Harper JW, Hannon GJ, Elledge SJ. Cancer proliferation gene discovery through functional genomics. Science 2008; 319: 620-624
26. Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with Ras oncogene. Cell 2009; 137: 835-848
27. Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM, Schuldiner M, Weissman JS, McManus MT. Rapid creation and quantitative
monitoring of high coverage shRNA libraries. Nature Methods 2009; 6:443-445
28. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 1974; 3: 355-359
29. Anderson SN, Towne DL, Burns DJ, Warrior U. A high-throughput soft agar assay for identification of anticancer compound. J Biomol Screen 2007; 12: 938-945
30. Thierbach R, Steinberg P. Automated soft agar assay for the high-throughput screening of anticancer compounds. Anal Biochem 2009; 387: 318-20
31. Maffia AM, Kariv I I, Oldenburg KR. Miniaturization of a Mammalian Cell-Based Assay: Luciferase Reporter Gene Readout in a 3 Microliter 1536-Well Plate. J Biomol Screen 1999; 4: 137-142
32. Pfeifer MJ, Scheel G. Long-term storage of compound solutions for high-throughput screening by using a novel 1536-well microplate. J Biomol Screen 2009; 14: 492-498.
33. Liu Y, Lacson R, Cassaday J, Ross DA, Kreamer A, Hudak E, Peltier R, McLaren D, Muñoz-Sanjuan I, Santini F, Strulovici B, Ferrer M. Identification of small-molecule modulators of mouse SVZ progenitor cell proliferation and differentiation through high-throughput screening. J Biomol Screen 2009; 14: 319-329
34. Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, Cetin C, Sieckmann F, Pau G, Kabbe R, Wünsche A, Satagopam V, Schmitz MH, Chapuis C, Gerlich DW, Schneider R, Eils R, Huber W, Peters JM, Hyman AA, Durbin R, Pepperkok R, Ellenberg J. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010; 464: 721-727
35. Mullenders J, Bernards R. Loss-of-function genetic screens as a tool to improve the diagnosis and treatment of cancer. Oncogene 2009; 28: 4409-4420
36. Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J, Sage D, Slisz J, Tran M, Straub C, Ramsey T, Iourgenko V, Huang A, Chen Y, Schegel R, Labow M, Fawell S, Sellers WR, Zawel L. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res 2007; 67: 11493–8
37. Lu J, Bai L, Sun H, Nikolovska-Coleska Z, McEachern D, Qiu S, Miller RS, Yi H, Shangary S, Sun Y, Meagher JL, Stuckey JA, Wang S. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res 2008; 68: 9384-9393
38. Petersen SL, Peyton M, Minna JD, Wang X. Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci USA 2010; 107: 11936-41191
39. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nature Cell Bio 2005; 7:591-600
40. Androic I, Krämer A, Yan R, Rödel F, Gätje R, Kaufmann M, Strebhardt K, Yuan J. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer 20089; 8: 391
41. George J, Banik NL, Ray SK. Bcl-2 siRNA augments taxol mediated apoptotic death in human glioblastoma U138MG and U251MG cells. Neurochem Res 2009; 34: 66-78
42. Irons E, Lord CJ, Ashworth A. Parallel RNAi and compound screens identify the PDK1 pathway as a target for Tamoxifen sensitization. Biochem J 2009; 417: 361-370
43. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Fröhling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009; 462: 108-112
44. Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, Döhner K, Bullinger L, Sandy P, Boehm JS, Root DE, Jacks T, Hahn WC, Gilliland DG. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009; 137: 821-834
45. Huang Y, Bao Y, Peng W, Goldberg M, Love K, Bumcrot DA, Cole G, Langer R, Anderson DG, Sawicki JA. Claudin-3 gene silencing with siRNA suppresses ovarian tumor growth and metastasis. Proc Natl Acad Sci USA 2009; 106: 3426-3430
46. Agarwal R, D’Souza T, Morin PJ. Claudin-3 and claudin-4 expression in ovarian epithelial cells enhances invasion and is associated with increased matrix metalloproteinase-2 activity. Cancer Res 2005; 65: 7378-7385
47. Castanotto D, Rossi J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009; 457: 426-433
48. Izumiya M, Okamoto K, Tsuchiya N, Nakagama H. Functional screening using a microRNA virus library and microarrays: a new high-throughput assay to identify tumor-suppressive microRNAs. Carcinogenesis 2010; 31: 1354-1359
49. Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y, McCue PA, Quong AA, Lisanti MP, Pestell RG. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci USA 2010; 107: 8231-8236
50. Palmer RD, Murray MJ, Saini HK, van Dongen S, Abreu-Goodger C, Muralidhar B, Pett MR, Thornton CM, Nicholson JC, Enright AJ, Coleman N; Children’s Cancer and Leukaemia Group. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res 2010; 70: 2911-2923
51. Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Chang K, Khan A, Leslie C, Parker J, Paddison P, Tam W, Ferrando A, Wendel HG. Genome wide RNA mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukemia. Nature Cell Bio 2010; 12: 372-379
About the Authors
Jason Borawski
Jason Borawski obtained a BS degree in Biology from Florida Gulf Coast University. He began his career working in the Gastrointestinal Cell Biology Division at Children’s Hospital in Boston, MA where he researched the role of K+ and Cl- conductances in intestinal epithelial biology and on receptor mediated intestinal mucosal immunity. In 2004, Jason joined Novartis Institutes for Biomedical Research in Cambridge, MA. Over the past six years, his work has focused on identification of target cellular host factors and pathways implicated in the progression of human disease. He has published methods for optimising siRNA transfection in vitro that have been critical in the assay development of high throughput genomic screening at Novartis. Jason has extensive experience utilising and exploiting RNAi technology and his work has contributed directly to the drug discovery pipeline at Novartis. His RNAi screening efforts have identified and validated novel drug targets involved in viral replication, apoptosis and tumour progression.
Contact the Author
email: [email protected]
Alex Gaither
Alex Gaither obtained his PhD from the University of Missouri-Columbia, focused on the cloning and molecular characterisation of zinc transport proteins. Alex went on to join Novartis Pharmaceuticals in Summit, NJ for a presidential post-doctoral fellowship in the Functional Genomics department. He worked for several years looking to identify genetic modulators of Alzheimer’s disease. He was then hired in 2001 by the Novartis Institutes for Biomedical Research as a biology lab head in the Functional Genomics department to focus on assay development and screening using RNAi technologies. In 2003, Alex was recruited to Novartis Institutes for Biomedical Research in Boston, MA to continue his work on RNAi screening in the Oncology and Virology disease areas. Since 2003, Alex has grown at Novartis into a Senior Investigator in the Developmental and Molecular Pathways where his group is focused on target ID and validation for novel therapeutics in Oncology.
Contact the Author
email: [email protected]
