

UV-Vis scan-based quality control of assay-ready plates for improved data integrity
Posted: 19 April 2011 |
Quality biological data requires both a high quality assay and a high quality compound. While assay quality is very closely monitored and has been intensively studied in the past, the quality of the final compound solutions being tested in an assay has received little attention. Quality of these samples is critical to the screening process, especially for XC50 determinations used in structureactivity relationship (SAR) analyses. Many laboratories have implemented routine analytical quality control (QC) on the stock solutions of all compounds, as well as quality assurance (QA) procedures for the weighing and liquid handling instrumentation used to produce the final assay-ready plates. Unfortunately, the stock sample QC and instrumentation QA together do not address the issues of assay plate production; indeed, stock sample QC does not address what happens to the sample once it has entered a compound management solution store at all…
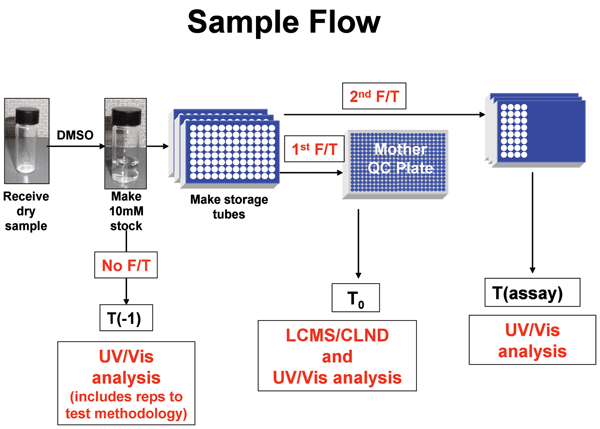
Figure 1 Process to obtain UV Scans of standards and assay samples. Solid compound samples are weighed
Quality biological data requires both a high quality assay and a high quality compound. While assay quality is very closely monitored and has been intensively studied in the past, the quality of the final compound solutions being tested in an assay has received little attention. Quality of these samples is critical to the screening process, especially for XC50 determinations used in structureactivity relationship (SAR) analyses. Many laboratories have implemented routine analytical quality control (QC) on the stock solutions of all compounds, as well as quality assurance (QA) procedures for the weighing and liquid handling instrumentation used to produce the final assay-ready plates. Unfortunately, the stock sample QC and instrumentation QA together do not address the issues of assay plate production; indeed, stock sample QC does not address what happens to the sample once it has entered a compound management solution store at all.
As we know, there are discrepancies in dispensed samples due to the sample precipitation, degradation, to automation malfunction and human error, likely among others. With the recognition of these potential errors downstream of the stock sample QC process, we have undertaken an effort to evaluate and implement a simple means to increase the quality of compounds dispensed in the biological assay plate distributions.
In this paper, we describe a simple process based on attaining and analysing rapid UV/Vis scans of all compound distributions. With it, we have analysed the assay plate production process first for compounds in some selected kinase screens, and then for all SAR compounds. Errors due to compound concentration or identity on an assay plate were detected and eliminated, even when the source of error could not be identified. The process has the advantage of having enough throughput to match biological SAR throughput, and uses one of the simplest, most inexpensive and robust analytical detection methods: UV/Vis spectral scanning.
In recent years, analytical QC of any compound has become routine, both at the synthesis stage before a compound is even submitted to a compound management laboratory10-13, as well as in the initial (mostly) DMSO stock solution14-16. Most laboratories now have purity and concentration thresholds which must be met by all DMSO stocks to allow them to be processed for further screening9,17-19. All these advances in the quality of the compound solution stocks were made possible by developments in analytical instrumentation which enabled the necessary throughput, sampling and sensitivity12,20,21. Unfortunately, none of the current analytical solutions involving some form of separation by liquid chromatography are capable of keeping up with the screening throughput. Simply put, the reason is fairly clear: separation involves testing one sample at a time, while screening techniques involve testing/reading entire multiwell plates at any one time. To develop a QC process for all assay solutions, a plate-based method, as described here, is desirable.
As mentioned before, compound management is a complex process involving multiple steps. To add to the complexity, the format of various assay-ready plates differs from assay to assay and it can involve unique steps for each process. While one can and should perform rigorous QA on all instrumentation used to ensure optimum performance, it would be very impractical to implement a product QC at every (different) step of this process (Figure 1). The approach we have taken here is to QC the final product only, which takes into account all steps along the way and allows removal or flagging of failed samples prior to testing. The process developed uses rapid UV/Vis scans at two steps of the process: the initial solubilisation step (T0) and the final assay solution (Tassay). The process involves no separation and is based on a direct scan-to-scan comparison of T0 and Tassay. What makes this a valid approach is the fact that T0 is directly correlated with an absolute measure of quality performed on the same initial stock using Liquid Chromatography / Mass Spectrometry / Ultraviolet Visible spectroscopy (LC/MS/UV) for purity determination and ChemiLuminescent Nitrogen Detection (CLND) for concentration determination.
Figure 1: Process to obtain UV Scans of standards and assay samples. Solid compound samples are weighed into vials and dissolved in neat DMSO to 10 mM. These solutions are transferred to storage tubes and frozen. The storage tube is thawed, a sample is taken and analyzed for identity, purity and concentration by LC-MS and CLND. This sample is also used as the T0 UV/Vis sample. The storage tube is then refrozen. The storage tube thawed again, sampled and appropriately diluted for distribution to various target assays. Tassay UV/Vis spectra are taken from these diluted plates that are ready to be stamped into assay plates. For some compounds, an additional UV/Vis sample was taken prior to the first freeze (T-1) during these experiments to determine sample changes upon the first freeze.
A description of the process developed, the method development and validation approach, the data processing and analysis solution, as well as the impact of this process on the overall drug discovery process will be described. Methods Compound distribution and dilution Figure 1 shows the process for generating samples and UV/Vis scans for both control and assay samples. In all cases, samples were weighed and dissolved in neat DMSO to 10 mM. (At this point, a pre-freeze/thaw sample, T-1, was taken and its spectrum obtained.) The remaining sample was placed into compound handling storage at 4°C. The DMSO solutions froze within minutes of entering the store. Shortly thereafter, the sample was thawed, and the control (T0) sample was cherry-picked using a Tecan instrument into a multi-well plate and its spectrum obtained. At this time, an absolute standard was also obtained. This sample was referred to Analytical Chemistry, and its structure, purity and concentration at this T0 time were confirmed by LC/MS/UV and CLND, respectively.
The sample was placed back into storage at 4°C where the solutions refroze. When a biological assay of the compound was requested, the sample was again thawed and an aliquot was cherry-picked using a Tecan instrument into ‘mother’ plates, and diluted in DMSO to desired starting concentrations. These ‘mother’ plates were subsequently serially diluted in DMSO in preparation for XC50’s replication. After removal of samples for assay, a 10 μl aliquot of the top mother plate concentration (typically 1 mM) was transferred into a UV clear plate (Greiner Bio-One, product #781801, UV-Star®) and diluted to 100 μl with DMSO for the assay UV/Vis scan (Tassay). (T-1 and T0 were similarly prepared and diluted to the same nominal concentration as Tassay).
UV/Vis scan production
UV/Vis scans were obtained with a 384Plus Molecular Devices Spectramax. All scans were taken in UV clear plates. All samples were scanned from 270 to 500 nanometres, at 10 nanometres. Typical compound spectra are shown in Figure 2A.

Figure 2: Detection and analysis of spectra differences between T0 and Tassay samples by UV/Vis spectroscopy. A. Typical UV/Vis spectral comparisons to determine quality of assayed compound solution (Tassay) vs standard (T0). Spectra were produced as described in Methods. The upper panel shows spectra of T0 and Tassay replicates of CMPND 2, which had correct identity and concentration in all samples. The lower panel shows spectra of T0, Tassay and retest samples of CMPND 3, which exhibited decomposition after T0. B. Plot of absorbances at all wavelengths for assay sample (Tassay) vs standard (T0). This plot enables quantitative comparison of differences between the solution of the compound that was assayed on the target and the standard. The upper panel shows a replot of absorbance values from 2A, upper panel, for CMPND 2 showing that slope and R2 approach 1.0 for a compound with correct identity and concentration in all samples. The lower panel shows a replot of absorbance values from 2A, lower panel, for CMPND 3 and that R2 is less than 1.0 for spectra indicating structure differences between the T0 and Tassay sample(s).
Analysis of UV/Vis spectra: Spectra of T-1, T0 and Tassay samples were compared quantitatively by plotting the absorbance of one spectrum vs. the other at all wavelengths (Figure 2B). The slope and R2 of this linear plot are each 1.0 if the two samples are identical in composition and concentration. If the concentrations differ, the slope deviates from unity; thus a slope of 0.5 indicates that the T0 was twice as concentrated as the Tassay, and a slope of 2 indicates that the Tassay was twice a concentrated as T0. If the spectral shapes differ, the R2 decreases. Typically R2 ≤ 0.95 indicates that the two compounds are different. We have found, however, that this parameter is conservative, and visual inspection was generally required for confirmation of structural differences. Of course, UV/Vis spectra cannot detect all structural differences, such as some ‘R’ group changes within a chemical series, but it does detect many structural differences and it detects all concentration errors for all compounds absorbing within the scanned range.
LC/MS/UV and CLND
Concentration was determined by Chemi – Luminescence Nitrogen Detection (CLND) which is an equimolar detection method that determines concentration based on the number of nitrogens present in a sample.
Flow-inject CLND method: Equipment used: Antek 8060 CLND, HTS CTC PAL autosampler, a Waters Corp. 1515 HPLC pump, and a Waters Corp. 996 PhotoDiode Array detector (PDA)*. No HPLC column was present. Run time: 1.5 minutes, Injection volume: 2μL, Flowrate: 0.2mL/minute 100 per cent methanol, PDA: 210- 400 nanometre scan, 1.2 nanometre resolution.
Prior to each plate of compounds analysed, a blank injection of DMSO was made. The instrument was calibrated with three to 100 mM caffeine standards in DMSO. Each sample was analysed in triplicate. Caffeine standards were analysed before and after test plates for system suitability.
Once concentration had been determined by flow-inject CLND, the samples were analysed by UPLC®-MS, a hybrid liquid chromatography and mass spectrometer method, for identity and purity. Waters Acquity SQD mass spectrometer, Waters Acquity PDA, sample manager (autosampler) and binary solvent manager were used for analysis. Column: Acquity UPLC® BEH C18, 1.7μm, 2.1×50 millimetres. Run Time: 1.50 minutes. 1.2 nanometre resolution; SQD scanning 100-1000 amu. Column Temperature: 60°C. Injection volume: 0.5μL. UPLC Pump Flowrate: 0.9 mL/minute. Solvent A: Water + 0.1 per cent HCOOH. Solvent B: ACN + 0.075 per cent HCOOH.
For each plate of compounds analysed, a standard test mixture (comprised of phthalates and other components) was analysed to ensure the instrument was fit for purpose then single injections of each compound was made. Injections of the standard test mixture were made every 80 samples.
Results
All SAR compounds at GSK go through a time sensitive potency determination (‘immediate processing’). The UV-based QC process described here is being optimised for all SAR compounds; however, the method and process were developed on the kinase compounds.
Kinases have a conserved ATP binding pocket22. As such, inhibitors for one kinase are often inhibitors for another kinase. Therefore, to enable rapid cross fertilisation between kinase programmes, most kinase SAR chemistry at GSK has been screened in ‘immediate processing’ as ‘all vs. all’. That is, all new chemistry for any kinase is screened up front against all other kinase assays running.
All vs. all is accomplished by providing each kinase assay with a replicate of a single compound cherry-pick and subsequent dilution. This parallel distribution increases certainty that all assays see the same compounds and concentrations and increases the validity of selectivity comparisons. To further increase data integrity, replicate assays are performed on a second (or third, etc) separate compound distribution. This allows a true test of the entire screening system in each replicate.
Typical replicate variability for our kinase SAR assay within this production system is well within 0.3 log units (twofold). However, we do observe catastrophic discrepancies in the data beyond that expected by the routine variance. Table 1 on page 50 shows a case of multiple replicate PDK1 assays of compound 1. The first six replicate PDK1 assays yielded an average pIC50 of 6.93 with a standard deviation (SD) of 0.1, but subsequent replicates increased the average pIC50 to 7.23, and the SD to 0.45. Importantly, in parallel we observed similar poor reproducibility in assays of this compound on other kinases in this compound distribution (not shown). To us, this suggested a difference in what was dispensed into the mother plate from the stock solution since assay plates generated in parallel as in Figure 1 on page 49 would be susceptible to this type of parallel error.
Compound decomposition, a dispense of the wrong compound or the wrong concentration into the ‘mother plate’ from which all assay plates in the distribution will be stamped in parallel, would lead to the parallel assay discrepancies as in Table 1. However, because this error was noticed in all parallel assays, it suggests that it is not derived from errors in replicating the mother plate into assay plates. Replication errors would lead to stochastic assay discrepancies in one assay only, not all.
| Table 1: Replicate PDK1 Data* and Absorbance at 300 nm for Cmpnd 1 assay samples | ||||||||||||||
| Replicate number | Replicate data | Average of all replicates | SD of all replicates | Average for data subsets | Std Dev for subsets | Replicate data | Average of all replicates | Std Dev of all replicates | ||||||
| pIC50 | A300 | pIC50 | A300 | pIC50 | A300 | pIC50 | A300 | pIC50 | A300 | pIC50 corrected for A300 | ||||
| 1 | 6.79 | 0.097 | 7.23 | 0.47 | 0.45 | 0.556 | 6.93 | 0.096 | 0.09 | 0.003 | 7.89 | 7.96 | 0.140 | |
| 2 | 6.87 | 0.093 | 7.98 | |||||||||||
| 3 | 6.95 | 0.094 | 8.06 | |||||||||||
| 4 | 6.98 | 0.101 | 8.06 | |||||||||||
| 5 | 7.00 | 0.094 | 8.11 | |||||||||||
| 6 | 7.01 | 0.097 | 8.11 | |||||||||||
| 7 | 7.7 | 1.221 | 7.81 | 1.206 | 0.10 | 0.045 | 7.70 | |||||||
| 8 | 7.85 | 1.242 | 7.84 | |||||||||||
| 9 | 7.88 | 1.156 | 7.90 | |||||||||||
We sought a simple means to determine whether any of the assay variability in Table 1 on page 50 had derived from distribution process errors.
Approximately 97 per cent of compounds screened have a chromophore in the 270 to 500 nanometre region of the electronic spectrum. UV/Vis spectroscopy is a rapid, inexpensive, nondestructive technology capable of providing both quantitative and qualitative compound information on small sample volumes in 384 well formats. In addition, the technique has the potential for throughput to enable us to analyse all mother plates from our SAR campaigns.
Without an absolute standard, UV/Vis is not an absolute analytical method, but it can provide rapid sample to sample comparative information, which is what we desired.
As such, a system was established to run, store and compare UV/Vis scans on all mother plates from kinase screening compound distributions. The goal for the system was to provide us with the ability to determine if the same compound and concentration was screened in replicate assays. The system and resultant database could then be used to test compound stability, dissolution, distribution and assay errors.
Typical scan comparisons for replicate distributions are shown in Figure 2A. The upper spectra of Figure 2A shows UV/Vis scans for a series of distributions of the same compound (Tassay samples one through four) compared to the T0 sample for the compound. As can be seen, the compound has a UV/Vis wavelength maximum of ~310 nanometres. The first three Tassay distributions have both maximum absorbance and spectral shape nearly identical to the T0 sample, but the fourth Tassay has a maximum absorbance ~ 10 times lower than the T0 indicating that the assay concentration for Tassay 4 was ~ 10 fold lower.
The lower set of spectra of Figure 2A shows a T0 spectrum compared to a Tassay sample and to a retest sample. The T0 sample shows an absorbance maximum at ~ 360 nanometres, but neither the Tassay nor the retest samples exhibit this. The data indicate that a different compound from the T0 standard was distributed into the assay. The discrepancy could have resulted from a mis-distribution or decomposition. Since scan of the retest sample matched that of the Tassay sample, the analysis indicates that the compound had decomposed.
As can be seen from these two examples in Figure 2A, both spectral shape and absorbance intensity are readily recognisable and provide a clear indication of whether the same compound and concentration was delivered in the replicate biological assays.
We applied this type of spectral analysis to solutions used to produce the pIC50 data in Table 1. Figure 3 shows the UV/Vis Spectra for the samples assayed in Table 1. The spectral shapes for all samples were similar with an absorbance maximum at ~ 300 nanometres, but the absorbance was >10 times lower for the samples that produced the low pIC50 data. Table 1, column three shows the absorbance at 300 nanometres (A300) for these samples. Clustering the data by A300 dramatically decreased standard deviations for the pIC50s (columns 10 and 11 of Table 1). The absorbance values were then used to correct the pIC50 values (column 12). As can be seen, the standard deviation for the overall data set decreased from 0.45 to 0.14 pIC50 units. Subsequent CLND analysis showed that the stock solution used for the low pIC50 samples indeed had ~ 10 fold lower concentration, validating the UV/Vis data. The data show that the UV/Vis process provides a simple means for ongoing assessment of whether the same compound and concentration are delivered when the biological assay of a compound is repeated.

Figure 3-A

Figure 3: UV/Vis spectra for samples assayed vs PDK1. Spectra were produced as described in Methods. A. Full scale, background subtracted spectra showing absorbance differences between replicates 1-6 and 7-9. B. Expanded scale, background subtracted spectra for low absorbing PDK1 assay replicates 1-6 showing spectral shape identity between replicates 1-6 and 7-9. The data support structural identity of all replicates and concentration differences between replicates 1-6 and 7-9.
To automate the spectral comparisons and simplify interpretations, we implemented a plot of the absorbances at all wavelengths from 280 to 400 nanometres for compound replicate spectrum one vs. replicate spectrum two (Figure 2B). This plot is linear, and the slope and R2 values are 1.0 for the perfect spectral match. When the two spectra diverge, the slope and R2 values provide parameters for automated trapping. The slope of the line provides a measure of the concentration differences between distributions; that is, a slope of two indicates that the compound in spectrum one is twofold more concentrated than that in spectrum two. Typically, we flag concentration differences > twofold between distributions (slope >2 or < 0.5). The R2 value for the line serves as a reporter for compound identity between distributions. We use an R2 value < 0.95 to flag identity differences; however, this reporter is false positive sensitive, so these discrepancies do require visual inspection of spectra for confirmation.
Because electronic spectrum analysis does not have the power to be the sole information source for positive structure ID, we have used the UV/Vis changes to initiate a more in depth LC-MS and CLND analysis on the few samples found incorrect by UV/Vis. This follow up information has provided the absolute ID or concentration to enable removing incorrect data from the corporate screening database. Based on the success of finding and trapping errors and increasing data integrity for kinase SAR screening, we expanded the experiment to a three month test of the other targets screened at one GSK site. The goal of this experiment was to determine the frequency of these errors.
Our compound picking process requires a freeze-thaw step prior to analytical analysis and assay distribution, so for the T0 to represent the true process, we sample it after this freeze-thaw. To test for changes during this freeze-thaw step, we also took a ‘T-1’ (pre-freeze) sample immediately upon compound dissolution, prior to freezing. We also added another analysis sample to allow us to compare our UV/Vis T0 samples with an absolute standard. This sample was referred to as ‘LCMS/CLND’ and was taken along with the UV/Vis T0 sample. The sampling process for this experiment is shown in Figure 1.
We analysed 559 sample replicates built into the 3020 T-1 UV/Vis scans and found no replicate discrepancies by either identity (based on R2 threshold or visual spectral comparisons) or concentration (equal to or beyond twofold). These results provided confidence in the validity of the process and methodology. W
e next analysed 2343 UV/Vis spectra comparisons of T-1 and T0 samples; this should report on compound loss (usually precipitation) due to the initial freeze-thaw step. From the 2,343 comparisons, we found 23 discrepancies; 21 of these were concentration differences, and two were identity differences. These data indicate that 99 per cent of the compounds in the study were unaffected by the first freezethaw cycle.
The 3,560 T0 scans that we collected contained 40 T0 sample replicates. These 40 samples represented the preparation of duplicate independent stock sample vials each compound. Four of these independent preparations were found to have greater than twofold concentration differences between the replicates. We found no identity differences. To determine whether the UV/Vis analysis had correctly detected concentration differences in the replicates, we compared the T0 UV/Vis data with parallel data for 19 of the samples that also had LCMS/CLND data. The comparison showed that all of the UV/Vis and CLND concentration ratios were within twofold of one another, and most were much closer, as shown in Figure 4. This further increased our confidence of the ability of the UV/Vis method to detect and quantify relevant sample to sample variations.

Figure 4: Comparison of CLND and UV relative concentration data for T0 replicates. Absolute or relative concentrations were determined by CLND or UV/Vis, respectively, as described in Methods for the two T0 replicates of each compound. To enable comparison of the CLND data with the UV/Vis, the replicate concentrations were ratioed. For these ratios, the concentration of the first replicate was divided by that for the second for all compounds and by each method. The bar-graph showing the ratio data for CLND and for UV/Vis enable comparison of data from the two methods. As can be seen, analysis of T0 replicates compared well between the two methods.
We then determined the sample discrepancy rate between T0 and Tassay samples and between one Tassay and another (Tassay-1 vs Tassay-n). Table 2 shows a summary of all of the data. We found that samples exhibited differences in both identity and concentration, and empty wells were detected as well (no absorbance above background). The data revealed an overall discrepancy rate of ~ two per cent for T0 to Tassay and 1.6 per cent when all T0 to Tassay and Tassay1 to Tassay(n) discrepancies were combined.
Table 2: Discrepancy Rate Analysis for all distributions
| Type of Sample Comparison | Number of spectra comparisons | Identity errors(%) | Concentration difference >2X (%) | Empty wells(%) | Sum of all errors(%) |
| T‑1 -> T0 | 2343 | 0.1 | 0.9 | 0 | 1 |
| T0 -> T0 | 40 | 0 | 10 | 0 | 10 |
| T0 -> T(assay) | 2428 | 0.8 | 0.95 | 0.2 | 2.0 |
| T(assay1) -> T(assayn)* | 1170 | 0.1 | 0.3 | 0.5 | 0.9 |
| Total T0 -> T(assay) + T(assay1) -> T(assayn) | 3598 | 0.6 | 0.7 | 0.3 | 1.6 |
To understand the source of these discrep – ancies, 43 samples representing T0 to Tassay discrepancies were re-picked, re-plated and subjected to UV/Vis and LCMS/CLND analyses. The goal for the experiment was to determine whether the discrepancies reflected true changes in the compound solutions or distribution errors between T0 and Tassay. Table 3 shows the output of this analysis. The 43 samples comprised 18 identity discrepancies, 20 concentration discrepancies and five apparently empty wells. We found that 16 discrepancies reproduced on retest and 27 did not. Both identity and concentration discrepancy samples reproduced for some samples and not for others (Table 3). None of the empty wells discrepancies reproduced.
Table 3: Reproducibility of discrepancies detected by UV/Vis scans
| Type of Discrepancy | Number of discrepanciesdetected in 1st analysis | Number of discrepancies that reproduced on retest | Number of discrepancies that did not reproduce on retest | Number of retest discrepancies that agreed with retest LCMS/CLND | ||
| Identity | 18 | 9 | 9 | 17 of 18* | ||
| Concentration | 20 | 7 | 13 | 19 of 20** | ||
| Empty wells | 5 | 0 | 5 | 5 | ||
| * UV detected one reproducible identity discrepancy not confirmed by LCMS | ||||||
| ** UV detected one reproducible 2-fold concentration loss not confirmed by CLND | ||||||
We interpret discrepancies that reproduced as indications of true changes (likely degradation) in the compound sample (as in Figure 2A), and we interpret discrepancies that did not reproduce as some type of issue with the initial sample. As such, the retest data indicate that the T0 to Tassay discrepancies we detected had derived from both sample degradations subsequent to T0 sample analysis and from issues with the initial sample.
Discussion
UV/Vis scan of compound distributions provides a simple means to enhance assay plate quality assurance and data integrity. From over five years experience with the technology, we find discrepancies in ~ two per cent of kinase compounds. In the broader, cross target class analysis we confirmed this rate and broke errors down to: 0.6 – 0.8 per cent mis-identity or decompositions, 0.7 – 0.95 per cent concentration errors and 0.3 – 0.5 per cent missed wells for all dispensed compounds.
Especially for multitarget parallel compound distributions, such as kinases, we have found that these can have major impact on data integrity and customer satisfaction. By using the data from the UV/Vis scans to find these errors, we are able to proactively address them.
Examples of the type of issues the UV/Vis scans have resolved for us include the following:
(i) Incorrect distribution volume from source plates for all compounds
(ii) Plate replication order error: Noting consistent differences in the pXC50’s for all compounds in a kinase distribution, the UV/Vis scans confirmed in one case that less compound had been dispensed than planned and in another plates were barcoded incorrectly. With this information, we were able to correct the concentrations or identity prior to posting
(iii) Confirmation or correction of selectivity data: UV/Vis spectral data were used to confirm that apparent selectivity seen for specific compounds were due to compound delivery differences, that is concentration differences rather than true potency differences
(iv) Detection of decomposition: The method has also enabled routine detection of standard or sample decomposition or precipitation in our DMSO storage solutions that result in loss of potency
(v) Conflicting data on compounds under consideration for progression to develop – ment have been resolved retrospectively by examining archival UV/Vis scans compared to new known standards
This work has been a close collaboration between our compound management and screening groups. As such, data implications have been discussed, and alternate means to alleviate issues discussed and implemented. One specific example of this type of impact on the data has been on ‘empty wells’. As indicated above, we found that a significant fraction of discrepancies come from missed cherry-picks from the tubes into the mother plate resulting in empty wells. To correct this, we now visually inspect mother plates for these empty wells and correct them prior to distribution.
We are currently working to completely automate and integrate the process directly into our compound distribution system. Our goal is to detect all sample discrepancies prior to compound distribution to the biological assay and to correct the discrepant samples prior to data analysis and posting.
References
1. Janzen WP, Popa-Burke I: Advances in Improving the Quality and Flexibility of Compound Management. J. Biomol. Screen. 2009, in press
2. Macarron R: Critical review of HTS in drug discovery. Drug Discovery Today 2006; 11(7-8):277-279
3. Mayr LM, Fuerst P: The Future of High-Throughput Screening. J. Biomol. Screening 2008; 13(6):443-448
4. Snowden MA, Green DVS: The impact of diversity-based, high-throughput screening on drug discovery: “Chance favours the prepared mind”. Curr. Op. Drug Disc. & Dev. 2008; 11(4):553-558
5. Makarenkov V, Zentili P, Kevorkov D, Gagarin A, Malo N, Nadon R: An efficient method for the detection and elimination of systematic error in high-throughput screening. Bioinformatics 2007; 23(13):1648-1657
6. Taylor PB, Ashman S, Baddeley SM, Bartram SL, Battle CD, Bond BC, Clements YM, Gaul NJ, McAllister WE, Mostacero JA, Ramon F, Wilson JM, Hertzberg RP, Pope AJ, Macarron R: A Standard Operating Procedure for Assessing liquid Handler Performance in High-Throughput Screening. J. Biomol. Screen. 2002; 7(6):554-569
7. Quintero C, Rosenstein C, Hughes B, Middleton R, Kariv I: Quality Control Procedures for Dose-Response Curve Generation Using Nanoliter Dispense Technologies. J. Biomol. Screen. 2007; 12(6):891-899
8. Popa-Burke I, Lupotsky B, Boyer J, Gannon W, Hughes R, Kadwill P, Lyerly D, Nichols J, Nixon E, Rimmer D, Saiz-Nicolas I, Sanfiz-Pinton B, Holland S: Establishing Quality Assurance Criteria for Serial Diution Operations on Liquid Handling Equipment. J. Biomol. Screen. 2009, in press
9. Comley J: Compound Management in pursuit of sample integrity. Drug Disc. World 2005; 6(2):59-78
10. Ripka WC, Barker G, Krakover J: High-throughput purification of compound libraries. Drug Disc. Today 2001; 6(9):471-477
11. Isbell JJ, Zhou Y, Guintu C, Rynd M, Jiang S, Petrov D, Micklash K, Mainquist J, Ek J, Chang J, Weselak M, Backes BJ, Brailsford A, Shave D: Purifying the Masses: Integrating Prepurification Quality Control, High-throughput LC/MS Purification, and Compound Plating to Feed High- Throughput Screening. J. Comb. Chem. 2005; 7:210-217
12. Schaffrath M, von Roedern E, Hamley P, Stilz HU: High- Throughput Purification of Single Compounds and Libraries. J. Comb. Chem. 2007; 7:546-553
13. Guintu C, Kwok M, Hanlon JJ, Spalding TA, Wolff K, Yin H, Kuhen K, Sasher K, Calvin Pl, Jiang S, Zhou Y, Isbell JJ: Just-in- Time Purification: An Effective Solution for Cherry-Picking and Purifying Active Compounds from Large Legacy Libraries. J Biomol. Screening 2006; 11:933-939
14. Yurek DA, Branch DL, Kuo MS: Development of a System to Evaluate Compound Identity, Purity, and Concentration in a Single Experiment and Its Applications in Quality Assessment of Combinatorial Libraries and Screening Hits. J. Comb. Chem. 2002; 4:138-148
15. Popa-Burke IG, Issakova O, Arroway JD, Bernasconi P, Chen M, Coudurier L, Galasinski S, Janzen WP, Lagasca D, Liu D, Lewis RS, Mohney RP, Sepetov N, Sparkman DA, Hodge CN: Streamlined System for Purifying and Quantifying a Diverse Library of Compounds and the Effect of Compound Concentration Measurements on the Accurate Interpretation of Biological Assay Results. Anal. Chem. 2004; 76:7278-7287
16. Letot E, Koch G, Falchetto R, Bovermann G, Oberer L, Roth HJ: Quality Control in Combinatorial Chemistry: Determinations of Amounts and Comparison of the “Purity” of LC-MS-Purified Samples by NMR, LC-UV and CLND. J. Comb. Chem. 2005, 7:364-371
17. Schopfer U, Engeloch C, Stanek J, Girod M, Schuffenhauer A, Jacoby E, Acklin P: The Novartis Compound Archive – From Concept to Reality. Combi. Chem. HTS 2005; 8:513-519
18. Houston JG, Banks MN, Binnie A, Brenner S, O’Connell J, Petrillo EW: Case study: impact of technology investment on lead discovery at Bristol-Myers Squibb, 1998-2006. Drug Disc. Today 2008; 13(1/2):44-51
19. Yasgar A, Shinn P, Jadhav A, Auld D, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A: Compound Management for Quantitative High-Throughput Screening. J Assoc. Lab. Autom. 2008; 13:79-89
20. Koppitz M, Brailsford A, Wenz M: Maximizing Automation in LC/MS High-Throughput Analysis and Purification. J. Comb. Chem. 2005; 7:714-720
21. Ari N, Westling L, Isbell J: Cherry-picking in an Orchard: Unattended LC/MS Analysis from an Autosampler with >32,000 Samples Online. J. Biomol. Screen. 2006; 11(3):318-322
22. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S: The Protein Kinase Complement of the Human Genome. Science 2002; 298: 1916-1934
23. Biondi, RM, Cheung, PC, Casamayor, A, Deak, M, Currie, RA and Alessi, DR: Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000; 19, 979-988
