

Compressed gases: an important component of an environmental monitoring programme
Posted: 25 August 2017 | Claudio Denoya, Daniele Pandolfi, Frank Panofen | 5 comments
The quality attributes of manufactured pharmaceutical products include the physical, chemical, and microbiological characteristics of the raw materials, excipients and active pharmaceutical ingredient (API), as well as the final drug product (see Table 1). Absence of microbiological contamination is considered a critical quality attribute due to its potential to dramatically impact, directly or indirectly, the safety and/or the efficacy of the drug product.

A large proportion of products labelled as sterile are manufactured by aseptic processing rather than terminal sterilisation. Because aseptic processing relies on the exclusion of microorganisms from the process stream and preventing microorganisms from entering open containers during processing, product bioburden – as well as the bioburden of the manufacturing environment – are important factors governing the risk of unacceptable microbial contamination. The terms ‘aseptic’ and ‘sterile’ are not synonymous. ‘Sterile’ is derived from the Latin sterilis (unfruitful), meaning, in modern terms, free from living germs or viable microorganisms that have the potential to reproduce. In contemporary aseptic healthcare product manufacturing, ‘aseptic’ describes the process for handling sterilised materials in a controlled environment designed to maintain microbial contamination at levels known to present minimal risk.1 Therefore, the importance of adequate and effective microbiological controls cannot be overstated.2
Control of the environment in which pharmaceutical products are manufactured is a key element of Good Manufacturing Practice (GMP). Included in this control, the monitoring of microbial contamination is essential.3 Physical and microbial monitoring of manufacturing cleanrooms, restricted-access barrier systems (RABS), and isolators should include the components listed in Table 2, which summarises the major components of an environmental monitoring (EM) programme. Evaluating the quality of air, surfaces, personnel garments and behaviours in a cleanroom environment should start with a well-defined written programme, employing scientifically sound methods of sampling, testing, and data analysis.4
Compressed gases: a brief regulatory overview
As shown in Table 2, the testing and monitoring of compressed air and other process gases, such as gaseous and liquid nitrogen, oxygen, argon, and carbon dioxide, that come into direct contact with pharmaceutical drugs during the manufacturing process is vital to assuring the quality and safety of these products. Compressed air is a critical process parameter, whose variability has an impact on the critical quality attribute (see Table 1) and should therefore be monitored or controlled to ensure the process produces the desired quality.
Unlike the food industry, the pharmaceutical industry does not have a clear-cut guideline or regulation that specifically addresses compressed air quality requirements, testing frequency, or number of samples. The individual manufacturer is responsible for assessing the risk and the effect that a contaminated compressed air supply could have on the final product. The following is a list of three documents from the International Council for Harmonisation (ICH) general guidance on developing a process for the commercial manufacturing of a new pharmaceutical product:
- ICH Q8(R2) (Pharmaceutical Development)
- ICH Q9 (Quality Risk Management)
- ICH Q10 (Pharmaceutical Quality System).5
In addition, some national standards organisations, such as the Compressed Gas Association (CGA) and the National Fire Protection Association (NFPA), provide guidance documents for compressed air sampling. The FDA and EU GMPs also mention that compressed gases should have similar or better physical and microbiological quality than the cleanroom air located in the area where the gases are used. Additional references are the FDA 21 CFR 100. 40 (Avoiding contamination with food additives) and 21 CFR 211. 113 (Control of microbiological contamination).
Table 1: Definitions of quality attributes
Quality attribute | A physical, chemical or microbiological property or characteristic of a material |
Key quality attribute (KQA) | Potential to impact product quality or process effectiveness Associated analytical method |
Critical quality attribute (CQA) | Directly or indirectly impacts the safety or efficacy of a drug product |
Overall, the general methodology and requirements for compressed gases are described in ISO 8573. First released in 1991 and updated in 2001 and 2010, ISO 8573 is now an international standard relating to the quality of compressed gases and contributing to a particle-free environment in pharmaceutical production facilities, both downstream and upstream. The standard consists of nine separate parts, with Part 1 specifying the quality requirements of the compressed gas, and Parts 2-9 specifying the methods of testing for a range of contaminants. This standard provides a variety of purity classes that can be incorporated into a robust quality assurance plan for this critical utility. ISO 8573 consists of the following parts:
- Part 1: Contaminants and purity classes
- Part 2: Test methods for aerosol oil content
- Part 3: Test methods for measurement of humidity
- Part 4: Test methods for solid particle content
- Part 5: Test methods for oil vapour and organic solvent content
- Part 6: Test methods for gaseous contaminant content
- Part 7: Test method for viable microbiological contaminant content
- Part 8: Test methods for solid particle content by mass concentration
- Part 9: Test methods for liquid water content.
Part 1 outlines the required purity classes based on the concentration of particles (such as dirt, rust, pipe deposits) and level of impurities (compressor lubricant, wear particles, vaporized lubricant) (Table 3). Other tests are described in Parts 2 through 9.
ISO 8573 Part 7 specifies a test method for distinguishing viable, colony-forming, microbiological organisms and related components (eg, yeast, bacteria, endotoxins) from other solid particles that may be present in compressed air. One of a series of standards aimed at harmonising air contamination measurements, ISO 8573 Part 7 provides a means of sampling, incubating and determining the number of microbiological particles. The test method is suitable for determining purity classes in accordance with ISO 8573-1, and is intended to be used in conjunction with ISO 8573-4 when there is a need to identify solid particles that are also viable, colony-forming units.
Table 2: Components of an environmental monitoring programme
Type | Component | Characteristics | Purpose |
Physical | Total particulate of room or enclosure air | – Total inert and viable particles – Active sampling – Particle counter | – Quality of environment – Root-cause analysis for source of contamination |
Differential pressures, positive pressure from a critical area to adjacent area | – 10-15Pa difference – Sensor | Prevents entry of contaminant particles from adjacent to critical clean area | |
– Air changes – Unidirectional air flow | Sensor | Quality of environment | |
Temperature and relative humidity | Thermometer, hygrometer sensors | Product quality and personnel comfort | |
Microbiological | Viable counts of room or enclosure air | Microbial counts by active (impactors and others) and passive (settle plate) air sampling | – Quality of environment – Root-cause analysis for source of contamination |
Viable counts on personnel glove and garment surfaces | – Effective personnel training and hygiene – Microbial counts by contact plates and swabs | – Contamination risk from operators – Evaluation of aseptic techniques and practices | |
Viable counts on manufacturing facilities, equipment and instrument surfaces | Microbial counts by contact plates and swabs | Effectiveness of aseptic techniques, and cleaning and disinfection of area and equipment | |
Compressed Gases | Total particulates and microbial counts | Sampling of gas by adequate particle counter and microbial gas sampling impactor with decompression adapter | Evaluation of compressed gas quality – Should be of equal or better quality than air in the critical area into which the gas is introduced |
Others | – Any other materials and equipment that might produce a risk of contamination – Analysis of contamination trends | – | – |
The presence of viable microorganisms is verified by exposing an agar nutrient to the compressed air sample. Sampling for qualitative and quantitative assessment is also provided in Part 7. A slit-sampler – a type of impaction air tester – is used, together with the method given in ISO 8573-4. Isokinetic sampling of the air is carried out and reduced until it is within the range of the sampler, as identified by the manufacturer. Pressure is reduced to atmospheric conditions and flow measurements are performed to establish compatibility with the manufacturer’s recommendations, or in accordance with ISO 8573-4. Where the flow is known, the time for the exposure of the agar media to the compressed air sample is recorded. Part 7 does not specify limits for microbial contaminants, with USP <1116> or in-house limits typically used instead.
Microbial impactors with compressed gas kit
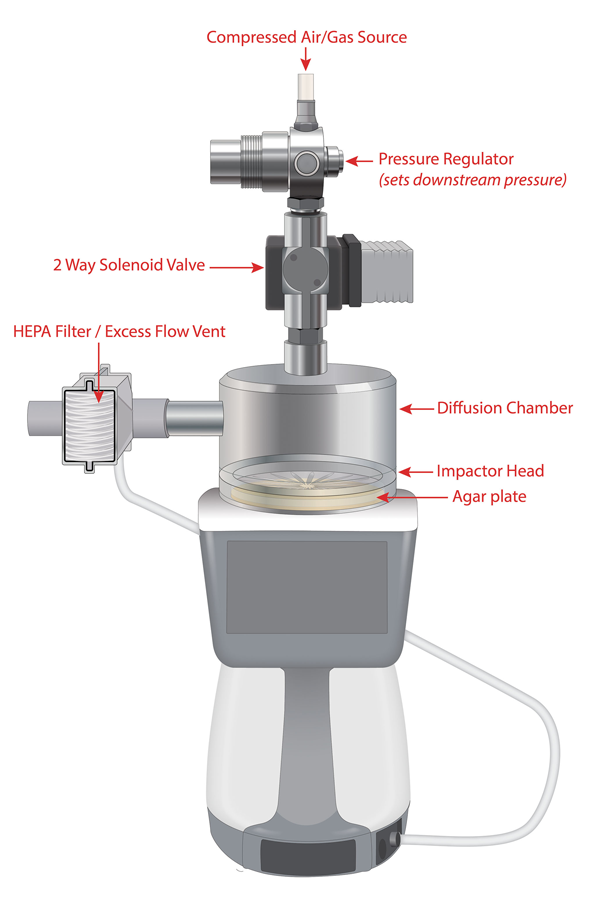
Figure 1: Example compressed gas setup
Compressed gas kits allow the microbial samplers to perform microbiological monitoring of compressed gases. Figure 1 shows an example setup. This compressed gas kit uses a special diffusion chamber mounted directly to the sampler inlet, the other end of which is connected directly to the high-pressure gas distribution lines.
The sampling is performed at balanced flow, where the compressed gas flow is equal to the sampling flow. The impactor inlet pressure coincides with the pressure of the compressed gas kit diffusion chamber.
For safety reasons – and to regulate the flow rate of the gas into the diffusion chamber – a pressure regulator is installed in the sampling line between the high-pressure gas source and the inlet to the compressed gas kit. A solenoid valve is connected downstream of the regulator and allows the sampler to synchronise the flow of gas with the start of a sample.
Instrumentation and selection criteria
One question users need to ask themselves is: When do I need to perform the decompression of the compressed gas? Some suppliers prefer decompression prior to sampling, and others promote sampling under compression with a subsequent decompression cycle. Gas for pharmaceutical purposes is decompressed before coming into contact with the product. Therefore, sampling after decompression is closer to the actual usage of the gas. In addition, there is no scientific evidence that a decompression of one order of magnitude down to ambient pressure will harm any microorganisms.6
There is no clear guidance from regulatory agencies on how to select an instrument. However, knowing that the decompression of the gas will have no influence on the viability of microorganisms, pharmaceutical manufacturers must select from a variety of instruments based on specific critical selection criteria:
- Size and weight
- Points of use
- Typically difficult to reach – mobile instruments/handhelds preferred
- Ease of use
- Software/data management capabilities.
Compressed gases need to be tested directly on-line, especially in critical areas. However, in designing a manufacturing line, monitoring points are not always established close to the point of use. Therefore, the connection points are often hard to reach and associated with a cleanroom setup. ISO 14698 states that sampling devices should be selected according to the area being monitored and take into account the effect of the sampling device on the process or environment being monitored.7
The instrument used for sampling must not contaminate the air that passes inside it, in order to avoid contamination of the cleanroom. A contribution to the particle load of the room is unacceptable, as it may create unpredictable events. Another aspect to be considered concerning the effect on the environment is the ability of the instrument to be properly cleaned and disinfected for cleanroom usage.
Table 3: Solid particulate, water and oil concentration per ISO class
ISO8573-1:2010 Class | Solid Particulate | Water | Oil | ||||
Maximum number of particles per m3 | Mass concentration mg/m3 | Vapour pressure dewpoint °C | Liquid g/m3 | Total oil (aerosol liquid and vapour) | |||
0.1-0.5 micron | 0.5-1 micron | 1-5 micron | mg/m3 | ||||
0 | As specified by the equipment user or supplier and more stringent than Class 1 | ||||||
1 | ≤ 20,000 | ≤ 400 | ≤ 10 | – | ≤ -70 | – | 0.01 |
2 | ≤ 400,000 | ≤ 6,000 | ≤ 100 | – | ≤ -40 | – | 0.1 |
3 | – | ≤ 90,000 | ≤ 1,000 | – | ≤ -20 | – | 1 |
4 | – | – | ≤ 10,000 | – | ≤ +3 | – | 5 |
5 | – | – | ≤ 100,000 | – | ≤ +7 | – | – |
6 | – | – | – | ≤ 5 | ≤ +10 | – | – |
7 | – | – | – | 5-10 | – | ≤ 0.5 | – |
8 | – | – | – | – | – | 0.5-5 | – |
9 | – | – | – | – | – | 5-10 | – |
X | – | – | – | > 10 | – | > 10 | > 10 |
Connecting an instrument to any sampling point in the gas line can become challenging as the accessibility is often very low. The size and weight of instrument will be a major consideration factor. Lifting heavy instruments to a sampling point close to the ceiling or pushing a huge enclosure below a filling line may become a tough exercise. Preferably, instruments should be flexible in usage and easy to carry and lift.
Instrumentation financials and return on investment
Aspects to consider when determining the right investment:
- Return on investment: rare measurement vs investment
- Multi-functional instruments vs dedicated instrumentation.
Monitoring compressed gases is not a frequent event in a facility’s environmental monitoring programme. Monitoring is typically performed during the first classification of the cleanroom. After the initial test, the test should be run monthly for six months, and if these results are satisfactory, the test should be performed quarterly for the rest of the first year. After the first year – and if the results are still satisfactory – the frequency can be lowered to twice per year in an ISO 5 area, corresponding to each cleanroom verification performed every six months in Europe, but reduced with good justification to once per year in the US.
Testing is not frequent, and investing in dedicated equipment with associated costs like validation, maintenance and repair, is difficult to justify financially. However, it is a mandatory regulatory requirement to monitor gases in facilities, with two optional strategies to consider.
One option is to use a service provider who routinely collects the required measurements. Scheduling the sampling and keeping control of the data and measurements can become difficult if there is not a good quality agreement in place.
The more favourable solution is a multipurpose instrument that can be used for other environmental samplings. The instruments are available in-house and need to be kept in a validated stage with an associated repair and maintenance scheme; the only additional costs are those of the accessories needed for sampling.
About the authors
 FRANK PANOFEN, PhD, is Sterility Assurance/Microbiology Product Line Manager, at Particle Measuring Systems. Dr Panofen has a Diploma in Chemistry from the University of Bielefeld and a PhD in molecular and cell biology from the University of Osnabrück. He has been an invited speaker at international conferences including ECA and PDA, with a strong regulatory background in pharmaceuticals. Dr Panofen is a certified Microbiological Laboratory Manager from ECA.
FRANK PANOFEN, PhD, is Sterility Assurance/Microbiology Product Line Manager, at Particle Measuring Systems. Dr Panofen has a Diploma in Chemistry from the University of Bielefeld and a PhD in molecular and cell biology from the University of Osnabrück. He has been an invited speaker at international conferences including ECA and PDA, with a strong regulatory background in pharmaceuticals. Dr Panofen is a certified Microbiological Laboratory Manager from ECA.
 DANIELE PANDOLFI is Global Product Specialist, Aerosol, Life Science Division, at Particle Measuring Systems. Mr Pandolfi has over 11 years’ experience in particle counter instrumentation and cleanroom contamination control. He has been responsible for the EMEA Service Department for the last four years and recently moved into the Global Life Sciences organisation.
DANIELE PANDOLFI is Global Product Specialist, Aerosol, Life Science Division, at Particle Measuring Systems. Mr Pandolfi has over 11 years’ experience in particle counter instrumentation and cleanroom contamination control. He has been responsible for the EMEA Service Department for the last four years and recently moved into the Global Life Sciences organisation.
References
To view references, please visit: www.europeanpharmaceuticalreview.com/4-17-Panofen
Issue
Related topics
Cleanrooms, Drug Manufacturing, Environmental Monitoring, Lab Equipment, QA/QC, Regulation & Legislation




Hi. I just want to ask if you can give acceptable parameters against dust, oil and water of the compressed air that is blown directly to product packaging material.
Literature shared on this website is knowledge enhancing and brain storming.
Production gasses required to be sterile when feeded on to Bioreactors in vaccine manufacturing?
What is the Microbial limit for the compressed gases monitored?
any guideline please.
what the frequency for the microbiological test for gases during sterile manufacture process?
For the Monitoring frequency of compressed gases after the initial test, the test should be run monthly for six months, and if these results are satisfactory, the test should be performed quarterly for the rest of the first year. After the first year – and if the results are still satisfactory – the frequency can be lowered to twice per year in an ISO 5 area, corresponding to each cleanroom verification performed every six months in Europe, but reduced with good justification to once per year in the US.
Could you please provide any reference or guidelines that support this programe?