

How the new draft of Annex 1, Manufacture of Sterile Medicinal Products impacts environmental monitoring programmes
Posted: 12 September 2018 | Daniele Pandolfi, Frank Panofen | No comments yet
This article explores how the new draft of Annex 1, Manufacture of Sterile Medicinal Products impacts environmental monitoring programmes.

Most of the relevant principles of the ISO 14644-1:2015 standard2 are included in the new Annex 1’s Chapter 5.26. The minimum requirements needed to classify a cleanroom, including the initial number of sampling sites and the required sample volume in critical zones (Grade A and B) are now present in Annex 1. All further decisions must be based on process knowledge and risk assessment. Consequently, it will become difficult to justify why lesser parameters for the qualification are chosen, especially for inspection of manufacturing environment sampling. This ties in deeply with the statement in Chapter 5.28, where “Clean room qualification (including classification) should be clearly differentiated from operational process environmental monitoring”. A clear distinction needs to be made between each phase of a clean environment’s lifetime.
This concept is represented in the equation shown in Figure 1. The classification of a cleanroom is based on particle load. There are no microbial limits given for this part of the process, but the revision contains a major change from the 2008 version.3
The classification of a cleanroom is based on particle load. There are no microbial limits given for this part of the process, but the revision contains a major change from the 2008 version.3
In chapter 5.25, particles of the size equal to or bigger than 5µm have been removed from the classification and qualification limit table for Grade A,but kept in the recommended limits for monitoring of the process environment.
The reasons for de-emphasising the 5µm limit in Grade A include:
- Harmonisation of the European Requirements with the recent release of ISO 14644-1, where the 5µm limit in ISO Class 5 has already been removed
- Sampling and statistical limitations for particles in low concentrations make it inappropriate
- Sample collection limitations for particles in low concentrations and sizes greater than 1µm make classification at this particle size inappropriate, due to potential particle losses in the sampling system.
De-emphasis of the 5µm limit refers only to the cleanroom classification process. The 5µm particles still represent an important indicator of possible contamination during the manufacturing process and, therefore, must be kept under control continuously during filling and manufacturing. Discrepancy in the treatment of 5µm particles between classification and monitoring are a foremost concern and may necessitate discussion on the possible risks of leaving out certain particle sizes during initial qualification. These sizes will still need to be within certain limits during monitoring.
The language pertaining to the responsibility of defining alert and action levels and limits has been made stronger and clearly refers to the cleanroom user, who must define the appropriate values based on a formal risk assessment and data trending analysis. This change emphasises regulators’ expectations that manufacturers set their action and alert limits based on historical data, process knowledge and a risk-based approach. In addition, it is important not only to define particle limits, but also an appropriate alarm strategy, which encourages the evaluation of ISO 14644-24 and its recommended practices.
The strategies set out in Figure 2 consider the importance of evaluating an alert or alarm situation using a series of events rather than a single spot value.
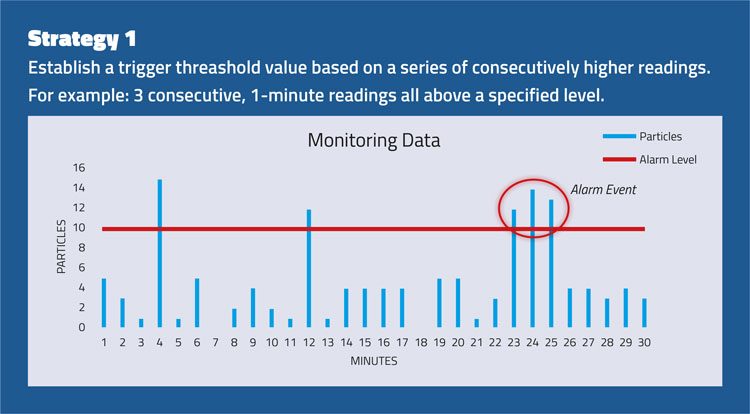
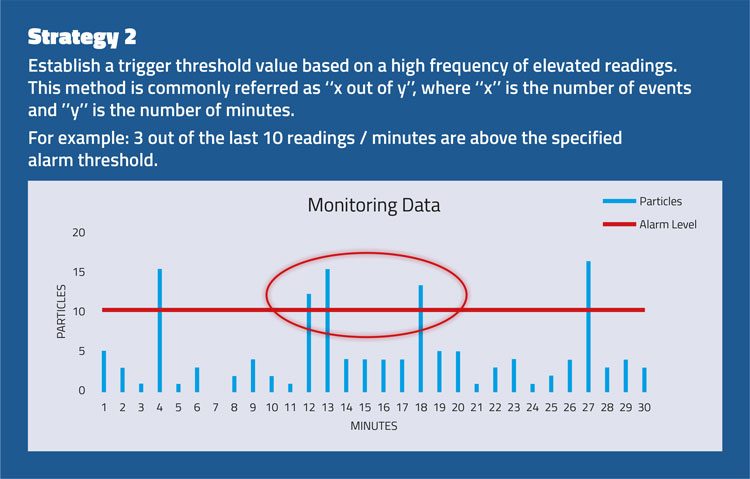
Figure 2
Requalification frequency
Paragraph 5.29 presents manufacturers with a challenge: bi-annual requalification of critical zones (Grades A and B) are becoming a standard of the industry. It is already a widespread practice, but many pharmaceutical companies have differing strategies that will need to be thoroughly explained in upcoming inspections. Modern technologies – including real-time methods for viable counts that minimise downtimes caused by the requalification process and therefore increase productivity – will become more crucial to the success of pharmaceutical companies.
It is interesting to note that throughout the new draft, Grade A and B environments are considered almost equal in the way they are treated for cleanliness.
Annex 1 and microbial impurities
Microbial impurities can be divided into “viable” and “non-viable” particles. “Non-viable” particles are inert and act as vehicles for viable particles, meaning they do not contain any microorganisms themselves. Laser-based particle counting, typically used for determining “non-viable” levels in a critical pharmaceutical environment, displays both “non-viable” and “viable” particles, which is comprised of “viable and culturable” and “viable but not culturable” (VBNC) impurities. The equation shown in Figure 3 represents what is seen by laser-based particle counting.

Figure 3
The components of this equation have widespread, and often incorrect, usage. Pharmaceutical cleanrooms are not classified using microbiological parameters, but by using non-viable / inert counts. Therefore, microbiological considerations start when the rooms are qualified for their intended use. As in the 2008 version, this is called the “in operation” stage and proposed action limits can be found in Table 2 of the draft. Although the values in the table look familiar, some major changes have been made:
- The values for Grade A zones are now set to 1 (was previously <1)
- No averaging of results is allowed.
Consequently, manufacturers are no longer allowed to ‘average out’ non-welcome results by looking at a scale of multiple measurements.
Each single result should be considered and cause a deviation resulting in a full investigation. However, this is true only for the qualification of the “in operation” stage and does not apply for the monitoring of the process. True routine “in operation” monitoring limits can be based only on historical data and locations, frequency, volume and duration of monitoring on a risk-based approach and data generated during the qualification, as stated in Chapter 9.5. This may create some confusion between the qualification stage’s “in operation” and the routine monitoring programme’s “in operation”.
Chapters 9.7 and 9.27 follow previously-established standards in terms of viable sampling, which can be found in other regulatory documents. In essence, sampling should be carried out as close as possible to the critical area in Grade A environments, but without posing any risk to the process and sampling itself. To do both has been a long-standing dilemma, and often requires a specialised approach using technologies such as single use.
The frequency of viable sampling has received an almost revolutionary renewal in the Annex 1 draft. Chapter 9.25 indicates that sampling must be frequent, and the combination of methods gives manufacturers ample control regarding which methodology and resulting data should be considered relevant for the sampling point. As always, the reasoning for all decisions must be documented and based on risk assessment and historical / scientific data. Interestingly, these strategies also apply to personnel monitoring (Chapter 9.26). Currently, manufacturers tend to avoid multiple samplings of operators in order to prevent contamination build-up and subsequent risk to the process and products. A possible solution could be the implementation of alternate sampling techniques, such as the use of swabs instead of contact plates.
One significant change is the recommendation for viable sampling to be performed continuously during routine process monitoring, as stated in Chapter 9.27. It will no longer be acceptable to have only small, snapshot sampling that does not characterise the entire manufacturing process. This concept was applied in the 2008 version for “non-viable” counts and has now been expanded into “viable” counts. Currently, continuous data generation can only be achieved by either real-time methods or long-term, traditional viable sampling that is quasi-continuous. The right combination of methods will become critical in the decision-making process.
Grade A and B zones are now considered almost equivalent in how they are treated from a monitoring perspective, and Chapter 9.33 imposes on manufacturers the need to identify all microorganisms found in these environments down to the species level. This new requirement emphasises:
- The importance of Grade B in final product quality
- The need for investigations in both cleanrooms
- The need for understanding the instruments used in these zones and their capability to contribute to contamination.
Conclusion
The consultation document for the new Annex 1 revision provides insight into upcoming regulatory trends. Significant emphasis is placed on manufacturers basing their decisions on an applied, risk-based approach. Monitoring plans should be proactively revised using growing knowledge of the process and risk assessment tools. The overall quality of products is sure to increase as a result of the released draft, with a stronger and deeper understanding of cleanroom performance.
REFERENCES
- Annex 1 draft version 2017, “Manufacture of Sterile Medicinal Products“. EU GMP Guide for Good manufacturing practice for drug products and drug substances.
- ISO 14644-1: 2015 – Cleanroom Classification Standard
- Eudralex Volume 4 – Annex 1 2008
- ISO 14644-2: 2015 – Cleanroom Monitoring Standard
BIOGRAPHY
 DANIELE PANDOLFI works for Particle Measuring Systems as Life Sciences, Aerosol Product Line Manager. He has worked in particle counter instrumentation and cleanroom contamination control for more than 10 years. While building strong customer relationships, he has helped many people solve their current Good Manufacturing Practice (cGMP) issues. Outside of work, he is a semi-professional photographer, an enthusiast of electronics, a lover of new technology, and a keen traveller.
DANIELE PANDOLFI works for Particle Measuring Systems as Life Sciences, Aerosol Product Line Manager. He has worked in particle counter instrumentation and cleanroom contamination control for more than 10 years. While building strong customer relationships, he has helped many people solve their current Good Manufacturing Practice (cGMP) issues. Outside of work, he is a semi-professional photographer, an enthusiast of electronics, a lover of new technology, and a keen traveller.

FRANK PANOFEN works for Particle Measuring Systems as Life Sciences, Product Line Manager. He has a Diploma in Chemistry from the University of Bielefeld and a PhD in molecular and cell biology from the University of Osnabrück. He has expansive experience in the field of applied pharmaceutical microbiology and serves as the Sterility Assurance / Microbiology Product Line Manager at Particle Measuring Systems. Frank has been an invited speaker at international conferences including ECA and PDA, with a strong regulatory background in pharmaceuticals. He is a certified Microbiological Laboratory Manager from ECA.





