

news
UK research consortium given £2 million to control monkeypox
£2 million will help fund a UK team of scientific experts to research monkeypox, uncover novel treatments and curb the spread of the virus.
List view / Grid view
£2 million will help fund a UK team of scientific experts to research monkeypox, uncover novel treatments and curb the spread of the virus.


The funding is being split across three research projects that will hopefully improve the treatment of COVID-19 and inform the development of vaccines and therapeutics.
A new cell therapy has been tested in patients with liver cirrhosis and found no significant adverse effects.


The results challenged the use of combined oral anticoagulant and antiplatelet therapy in patients with atrial fibrillation, especially those without an indication for antiplatelet therapy.
Researchers have shed light on how taking aspirin can help to prevent bowel cancer. They found that the painkiller blocks a key process linked to tumour formation...
Machine learning has detected one of the commonest causes of dementia and stroke, in the most widely used form of brain scan (CT), more accurately than current methods.


Taking painkillers during pregnancy could affect the fertility of the unborn child in later life, research suggests...


Drugs normally used to treat cancer could reduce the disfigurements of thousands of children born with life-threatening blood vessel defects...


Health Data Research UK is awarding £30 million funding to six sites across the UK to address challenging healthcare issues through the use of data science...


In this free-to-view Stem Cells In-Depth Focus, you can find out how computational ('in silico') methods can help to rationally choose bioactive small molecules to improve stem cell differentiation. The differentiation of pluripotent stem cells to hepatocyte-like cells is the focus of a second interesting article...

13 June 2013 | By Thierry Le Bihan, SynthSys and Institute of Structural and Molecular Biology, University of Edinburgh
In recent years, mass spectrometry (MS) based proteomics has moved from being a qualitative tool (used to mainly identify proteins) to a more reliable analysis tool, allowing relative quantitation as well as absolute quantitation of a large number of proteins. However, the developed quantitative methods are either specific for certain…
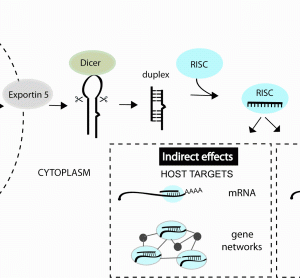
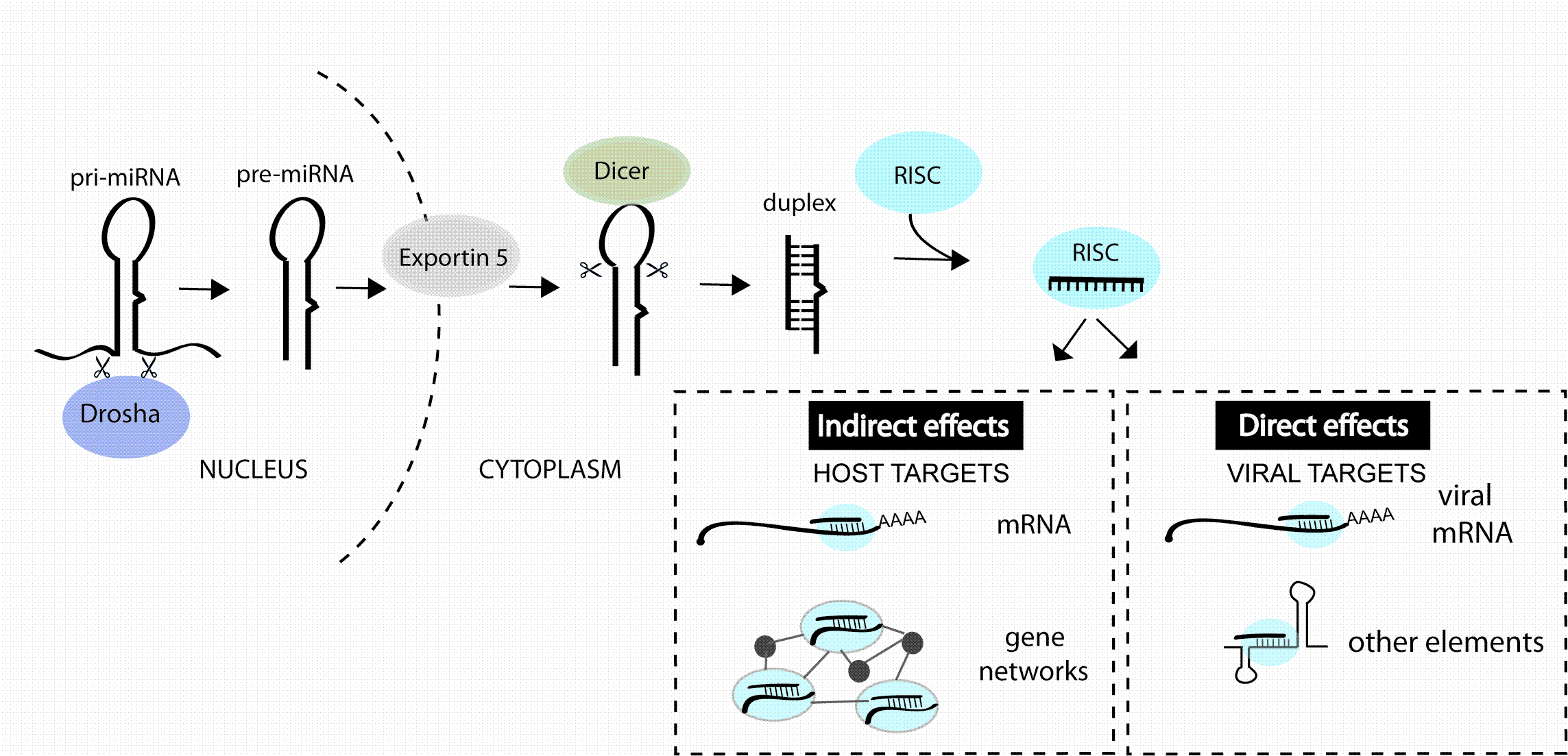
13 December 2011 | By Nouf N. Laqtom, University of Edinburgh & King Abdulaziz University and Amy H. Buck, University of Edinburgh
microRNAs (miRNA) are a class of non-coding RNA that regulate the precise amounts of proteins expressed in a cell at a given time. These molecules were discovered in worms in 1993 and only known to exist in humans in the last decade. Despite the youth of the miRNA field, miRNA…

22 February 2010 | By Peter Ghazal, Professor of Molecular Genetics and Biomedicine, University of Edinburgh and Head of Division of Pathway Medicine and Associate Director of Centre for Systems Biology, Edinburgh, Al Ivens, Head of Data Analysis, Fios Genomics Ltd and Thorsten Forster, Statistical Bioinformatician, Division of Pathway Medicine, University of Edinburgh
In conventional pharmacogenomic studies, genetic polymorphisms (including single nucleotide and copy number variations) are elucidated from case-control distribution of individuals usually representing ethnicity, severity of disease, and positive or negative response to treatment. However, the interpretation of a single genetic marker in this context is complicated, as the same marker…
