Quality by Design for generic products: opportunities and challenges
Posted: 22 February 2017 | Patrick Crowley, Sean McCrossen | No comments yet
The first part of an in-depth article on Quality by Design (QbD), published in European Pharmaceutical Review in December 2016, focused on designing quality in to a pharmaceutical drug product by considering dosage form design, and understanding the importance of active pharmaceutical ingredients’ (APIs’) and excipients’ properties.

In the second instalment of this two-part series, the possibilities for implementing manufacturing efficiencies, particularly those concerned with process monitoring and control, will be explored, thus providing the reader with the rest of the picture on how generics manufacturers might take advantage of QbD concepts in order to gain competitive edge.
Solid dosage form manufacture Yu et al provide a comprehensive listing of material and quality attributes, as well as processing parameters, that are relevant to operations for the manufacture of tablets (or capsules) and that merit programmes to characterise behaviour from a QbD perspective1. The following commonly used operations are considered here:
- Blending
- Size reduction (drug and/or granulated material)
- Granulation (size enlargement) or densification
- Drying (removal of water or other solvent)
- Compaction/compression (tablet or compact formation)
- Coating tablets or granules.
Such operations have historically been controlled by defining the equipment type, operating conditions and time of operation. However, variations in input materials (drug, excipients and process intermediates), scale of operation, environmental conditions and other factors, such as ‘wear and tear’ of equipment, could cause excessive (‘over’) or ‘under’-processing, thus compromising the attributes of output materials. Novel process analytical technologies (PAT) now promise to change processing paradigms by using online process monitoring capability with automatic termination of the process or sub- process when the requisite material attributes are attained. Possibilities associated with common process operations, or their subsets, are presented here as examples. They do not reflect the totality of emerging opportunities in a rapidly changing environment, particularly with respect to instrumentation for online monitoring. For instance, process analysers are mainly spectroscopic at present, but chromatography-based systems are also emerging. The potential for at-line, in-line or on-line measurements to monitor and assure quality can contribute to flexible and more reliable manufacturing operations and reduced energy consumption, as well as decreasing the need for end-product testing2.
Blending
Magnesium stearate is employed extensively as a lubricant in tablet manufacture. Many factors contribute to the variability of this otherwise excellent lubricant. Blend time is usually a critical process parameter. A blend time that is too long (‘over-lubrication’) leads to weak compact formation. It may also retard drug release. Under-lubrication generally causes die wall friction and tablet defects (‘sticking’ and ‘picking’).
Near-infrared spectroscopy (NIR) has been successfully utilised to monitor, on a continuous and non-invasive basis, the incorporation (blend homogeneity) of magnesium stearate in granule blends for tableting3. In one such study, spectra were captured for each rotation of the blender to monitor such homogeneity. Material was also sampled by conventional means (sample thief), dispersion homogeneity being determined by conventional analysis (atomic absorption spectroscopy). Figure 1 shows traces of standard deviations for lubricant distribution as a function of the number of blender rotations during a six minute time period (earlier sampling at 1.5 minutes/26 rotations showed that dispersion homogeneity did not meet United States Pharmacopoeia standards). The results correlated with homogeneity values determined by atomic absorption spectroscopy.
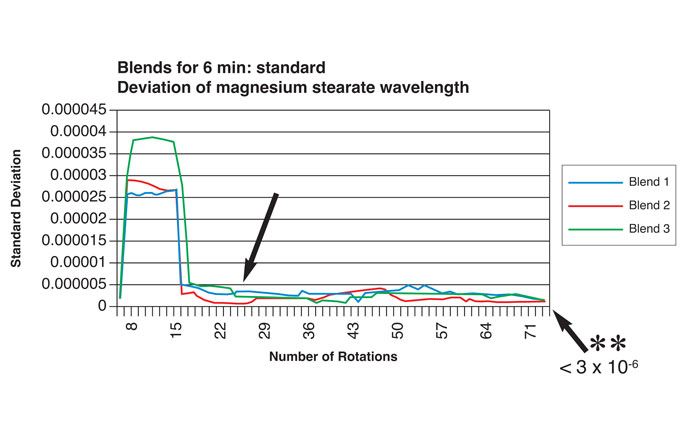
Figure 1: Relationship between number of blender rotations and magnesium stearate dispersion homogeneity by NIR analysis (reproduced with permission from reference 7)
A thermal effusivity technique has also been utilised to monitor magnesium stearate incorporation in a blend for tableting. When this measurement is constant, the relative standard deviation is minimal, indicating that the blend is homogeneous4. The technique requires that the sample be static for a brief period, requiring some form of ‘bypass’ in a continuously moving powder bed, or that the mixing process be briefly stopped.
In essence, PAT can play useful roles in optimising lubricant incorporation in blends for tableting, avoiding under- or over-blending that can compromise compaction and tablet quality. This could conceivably allow batch- or equipment-specific blend times, for instance, when equipment change is warranted due to changed off-take forecasts. The technique also has potential for qualifying alternative sources of lubricant where blend time might need to be modified for optimum lubrication.
Size reduction and enlargement
A drug may need to be size-reduced to improve its biopharmaceutical performance (e.g., micronisation), rendering a material physically more homogeneous (by breaking up lumps or aggregates) or to facilitate homogenous admixture with other components. Excipients may also, on occasion, need to be size-reduced (for instance, lactose in pulmonary inhalation products). Size enlargement processes (granulation) are also common, as in granule-forming operations. Process analytical techniques to continuously monitor particle size, be it reduction or enlargement, are considered together here for the sake of clarity, particle size being the common material attribute.
Laser diffraction methods based on estimates of mean particle diameters have been used to monitor particle growth (granule formation) during fluid-bed granulation or when coating fluidised particles. The profiles in Figure 2 illustrate the fall-off in small particle counts during processing as they are incorporated in larger (so-called ‘coarse’) granules. Excessive energy consumption is avoided, the process being terminated when the requisite size distribution is achieved.
Drying/solvent removal
The rate and extent of water or organic solvent removal during drying, granulation or coating can be affected by input variables such as temperature, air ‘dryness’, its velocity and the aerodynamics in fluidised systems (as well as time). Relative humidity of input air can vary with time of day, season and geographic location. Thus, output material can be ‘over’ or ‘under-dried’ if process conditions are defined in simplistic time and temperature terms. Inadequate drying could compromise the stability of moisture-sensitive materials while ‘over-drying’ could lead to brittle granules and attrition during subsequent processing. Probes that monitor the relative humidity or temperature of input and output air have historically been used to define the drying end point; a lack of difference between input and output air values being interpreted as indicating that moisture is no longer leaving the material so is effectively ‘dry’. However, such surrogate markers may not always be adequate as rate and extent of drying can be influenced by the aforementioned environmental factors as well as by material bulk density, affecting the rate of water vapour diffusion through the solid being dried. A measure of the ‘true’ moisture content may be required. NIR has the potential to provide real-time monitoring of moisture content, since water exhibits good absorption in the NIR spectral region. It has shown potential in fluid bed granulation5 .
The majority of APIs and excipients absorb NIR radiation. The technique is non-destruc – tive, non-invasive and rapidly generates data. As such it offers much potential for online monitoring of processing and quality attributes, such as blend uniformity or content uniformity in unit dosage forms. It may also be capable of determining the ‘spatial distribution’ of components in the finished product via an imaging technique (‘chemical imaging’)6 . Other vibrational spectroscopy techniques such as mid-infrared or Raman may also be valuable for online monitoring of processes, being non-destructive, speedy and non-invasive. Raman spectroscopy was shown to control tablet coating accuracy and homogeneity for a fixed dose combination product, where one of the drugs was spray-coated onto tablet cores. Content accuracy and uniformity were monitored online, with an automated process endpoint. The technology was robust and scalable from laboratory to industrial scale7 .
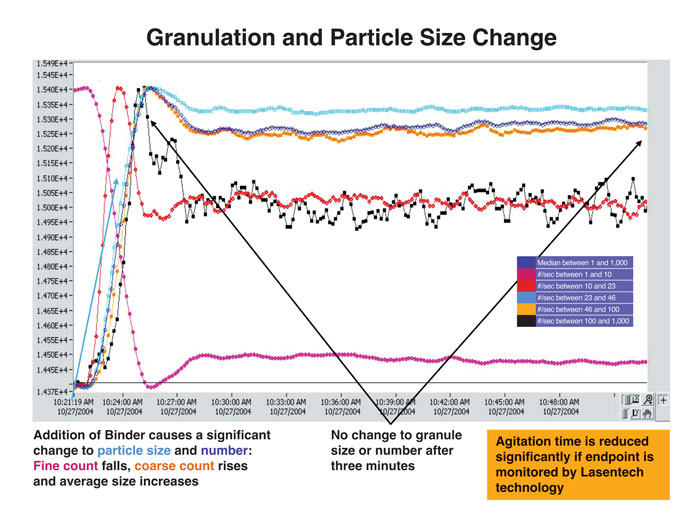
Figure 2: Granulation and particle size distribution (Lasentech) after addition of granulating agent
Where possible it could be advantageous to select excipients with spectroscopic characteristics that render them suitable for monitoring techniques such as NIR. Defining suitable process monitoring tests, process control and endpoint requirements are of prime importance during formulation and process development.
There are many design, operational and interpretational complexities associated with process analytical techniques and instrumentation. They may require significant investment in instrumentation, data collection systems, software development for setup and qualification, and for monitoring and maintenance programmes. Calibration and model development to qualify a technique tends to be material and process-specific. Nevertheless, cumulative experiences and advances in analytical instrumentation are likely to reduce complexity, increase robustness and refine interpretative capability. Regulatory agencies encourage investment in such technologies as a means of achieving better control of processes and products. Operational efficiencies, as well as product and process consistency, can benefit from such investments.
Compact formation

Figure 3: Benchtop compaction simulation equipment. Provided by and reproduced with permission of owners
Traditional paradigms concerning tablet compact formation involved monitoring hardness and compact weight, and possibly, friability ‘at line’, to ensure appropriate ruggedness and disintegration properties of the formed compacts. Nowadays, tablet presses are capable of automatic compaction to a defined pressure. If the compression properties of a formulation have been shown to be consistent it is possible to control output parameters such as hardness, friability and disintegration by the input parameter, compression force. However, such relationships can be tablet press-specific and depend on variables such as compression speed, angular velocity (being press size-dependent) and properties of excipients, such as residual moisture content or particle size or shape. Hence, compression behaviours merit periodic confirmation, particularly when qualifying a new source of excipient or active ingredient, or when compaction equipment is changed. Such diligence provides assurance of consistency of input/output relationships, or indicates where re-definition is necessary. Generating such information at large scale would be a demanding exercise.
Fortunately, the compaction process can be considered (in most cases) to be a ‘unit operation’. The advent and use of ‘benchtop compaction simulators now makes it possible to carry out many compression-related studies at laboratory scale. Compression cycles can be programmed to simulate different machine types and variables such as rotational speeds and centrifugal forces.
Ejection force could be monitored to qualify new sources of lubricating agents or different modes of incorporation in blends for compression. Figure 3 provides illustrations of equipment and references their providers.
Cycle time reduction in manufacture and testing
Monitoring and ‘control’ of quality, as previously (and mostly still) practised, can be unwieldy and slow. Monitoring ‘at line’ may mean interrupting the process so that ‘intermediates’ can be tested. Manufacture can accordingly comprise a series of ‘stop and start’ operations. In addition, GMP-based systems invariably require paperbased approval and many signatures. Overall approval times can greatly exceed manufacturing and testing times, even when there are no halts for testing intermediates. The timelines in Table 1 are not uncommon. Re-testing or reworking material prolongs cycle times. Yet, such monitoring and approval systems have done little to reduce product and process defects and recalls.
The QbD insights and PATs that have been briefly discussed here have the potential to provide more reliable processes and eliminate ‘stop/start’ modes of manufacture. This should facilitate the adoption of ‘continuous’ rather than batch manufacture. A well characterised process, with online ‘quality monitoring’ has the potential to greatly reduce cycle time. There is also much scope for innovative solutions to the ‘paper-based bottlenecking’ that contributes to the month- long (or greater) approval times in Table 1. Quality can be checked without interrupting production and aligns well with the concept of real-time release. Linking online monitoring data electronically to a batch review process, with an appropriate e-verdict on findings (or highlighting a potential issue), could substantially reduce review time, much of which is currently taken up by correcting documentation and other errors (with associated need to re-circulate for review and approval).
Table 1: Cycle Time for Manufacture and Approval*
| Activity | Time |
| Batch Manufacture | 1 day |
| Testing | 3 days |
Approval – ‘manual’ – 80 pages of records to review – sequential signatures |
40 days |
* privately-sourced information
There are many challenges to providing a verdict on overall quality, including analytical techniques, data collection, interpretation, and integration in a holistic way that integrates quality monitoring and control operations. Significant skill-sets and expertise are necessary. This will require substantial staffing as well as investments in facilities and equipment but the rewards can be substantial.
Conclusions
A systematic, science-based strategy for dosage form and process development, rather than an approach based on ‘copying’ the composition of an innovator product, can be a valuable investment in a generic drug development programme. It requires a strategic commitment to developing a QbD-based culture and a technical function resourced to develop the opportunities for efficiencies and savings.
Generics organisations experience time pressures for product filings and approvals. In the United States (depending on the type of abbreviated new drug application [ANDA] filed) a ‘first approval’ gains 180-day exclusivity; price competition then erodes margins. A well-conducted QbD programme, clearly communicated in ANDA filings, could reduce the risk of delayed approvals. It is important, therefore, that sufficient time and resource is allocated to the generation and communication of high-quality QbD information. Regulatory agencies welcome dialogue and discussion on plans and proposals for QbD-associated programmes based on good science. Key agencies also cooperate with each other on pilot projects in promoting the QbD way, such as the parallel assessment initiative.
In closing, it is hoped that some of the observations and thoughts in this two-part article help generics companies to consider appropriate investments with respect to QbD programmes and to reap the benefits of such commitment.
About the authors
 Patrick Crowley is a pharmacist by training and has spent his entire career involved in dosage form design, in the UK and USA. He made significant contributions, as Laboratory Scientist and Department Manager, in the development of 21 novel molecular entities to medicinal products. These included novel chemical entities, fermentation products and biopharmaceuticals. He also participated in and managed groups developing innovative line extensions of commercial products that secured intellectual property rights. Since retiring as a Vice President in Pharmaceutical Development at GlaxoSmithKline he has operated as an independent consultant and lectures as a Visiting Professor at The School of Pharmacy, Kings College, London.
Patrick Crowley is a pharmacist by training and has spent his entire career involved in dosage form design, in the UK and USA. He made significant contributions, as Laboratory Scientist and Department Manager, in the development of 21 novel molecular entities to medicinal products. These included novel chemical entities, fermentation products and biopharmaceuticals. He also participated in and managed groups developing innovative line extensions of commercial products that secured intellectual property rights. Since retiring as a Vice President in Pharmaceutical Development at GlaxoSmithKline he has operated as an independent consultant and lectures as a Visiting Professor at The School of Pharmacy, Kings College, London.
 Sean McCrossen is a chartered chemist and Fellow of the Royal Society of Chemistry. Sean has worked within the pharmaceutical industry for 30 years, specialising in analytical chemistry, chemical development and product support. Sean runs his own consultancy, specialising in CMC technical development and regulatory services to the pharma industry. He currently is working with worldwide pharma clients, regulatory affairs groups and CMO/CROs on both NCEs and generics.
Sean McCrossen is a chartered chemist and Fellow of the Royal Society of Chemistry. Sean has worked within the pharmaceutical industry for 30 years, specialising in analytical chemistry, chemical development and product support. Sean runs his own consultancy, specialising in CMC technical development and regulatory services to the pharma industry. He currently is working with worldwide pharma clients, regulatory affairs groups and CMO/CROs on both NCEs and generics.
References
- Yu L. Pharmaceutical Quality by Design: Product and Process Development, Understanding and Control. Pharmaceutical Research. 2008; 25 (4) 781-791
- Chen Z, Lovett D, Morris J. Process Analytical Technologies and Real Time Process Control. A review of some spectroscopic issues and challenges. Journal of Process Control, Elsevier. 2011
- Naicker K, Kilian G, Olivier J. Introducing PAT using NIR Analysis for a Pharmaceutical Blending Process. Pharma Manufacturing. 3rd May 2007
- Nakajawa H, et al. Real Time Monitoring of Lubrication Properties of Magnesium Stearate Using NIR Spectrometry and a Thermal Effusivity Sensor. Internat. J. Pharmaceutics. 2013; 441 402-413
- Sever NE, et al. Chapter 35 in Process Analytical Technology in Solid Dosage Development and Manufacturing: Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice. Elsevier. 2009
- Lyon RC, et al. Near Infrared Spectral Imaging for Quality Assurance of Pharmaceutical Products: Analysis of Tablets to Assess Powder Blend Homogeneity. AAPS Pharm Sci Tech. 2002; 3 (3) Art 17
- Wirges et al. Monitoring of an Active Coating Process for a Two-layer Tablet-model. Development Strategies. J. Pharm Sci. 2013; 102 (2) 556-566