

Conformational Bias: A key concept for protein kinase inhibition
Posted: 28 February 2012 |
Protein kinases act as molecular switches with remarkable plasticity and dynamics upon interaction with specific regulatory domains as well as modulators. Conformation provides a conceptual framework for understanding many aspects of kinase biology. The kinase domain has precise structural prerequisites for signal transfer and can oscillate between two major conformations: an on state with maximal kinase activity (active kinase) and an off state with minimal activity on the other extreme. Conformational bias, i.e. a shift in the equilibrium between active and inactive conformations is a key determinant in kinase regulation and can be brought about by many factors including post-translational modifications, regulatory proteins, ligand binding etc. Kinase inhibitors can be viewed as particular ligands to protein kinases. As the mode of action is linked to the binding mode, the selectivity as well as the kinetics of kinase inhibitors can often be rationalised based on the target conformation. Pathologic kinase deregulation often involves a shift towards the active conformation, leading to constitutive signalling. In this review, we discuss how mutations act as a conformational bias and depending on the mode of action can lead to activation of the protein kinase that can either result in resistance or contribute to the efficacy of kinase inhibitors. Deregulation of protein kinase activities by mutations and/or amplification are associated with a variety of pathologies ranging from cancer to inflammatory diseases, diabetes, infectious diseases and cardiovascular and metabolic disorders…
Protein kinases act as molecular switches with remarkable plasticity and dynamics upon interaction with specific regulatory domains as well as modulators. Conformation provides a conceptual framework for understanding many aspects of kinase biology. The kinase domain has precise structural prerequisites for signal transfer and can oscillate between two major conformations: an on state with maximal kinase activity (active kinase) and an off state with minimal activity on the other extreme.
Conformational bias, i.e. a shift in the equilibrium between active and inactive conformations is a key determinant in kinase regulation and can be brought about by many factors including post-translational modifications, regulatory proteins, ligand binding etc. Kinase inhibitors can be viewed as particular ligands to protein kinases. As the mode of action is linked to the binding mode, the selectivity as well as the kinetics of kinase inhibitors can often be rationalised based on the target conformation. Pathologic kinase deregulation often involves a shift towards the active conformation, leading to constitutive signalling. In this review, we discuss how mutations act as a conformational bias and depending on the mode of action can lead to activation of the protein kinase that can either result in resistance or contribute to the efficacy of kinase inhibitors.
Deregulation of protein kinase activities by mutations and/or amplification are associated with a variety of pathologies ranging from cancer to inflammatory diseases, diabetes, infectious diseases and cardiovascular and metabolic disorders1-4.
The success of imatinib, as a first in class kinase inhibitor, which was approved for the treatment of chronic myeloid leukaemia (CML) in 2001, triggered a worldwide effort in the pharmaceutical and biotech industry leading to the registration of 14 small-molecular weight KIs in the last decade (Table 1, below). These KIs target a handful of protein kinases, mainly for oncological indications and are, with the exception of the rapalogs, directed towards the ATP-site of these protein kinases. More than 150 kinase-targeted drugs are in clinical development and many more in various stages of pre-clinical development including phosphoinositide- 3 kinases (PI3Ks), mTOR, AKT and cell cycle regulating kinases3,5-8. The main challenges for kinase drug discovery today are (i) assessing the genetic linkage of the target kinase to the disease pathogenesis9-11, (ii) understanding the mechanisms of resistance to kinase inhibition12-14, (iii) patients stratification and finally (iv) identification and validation of novel kinase targets15-17.

A recent review of kinase mutations in human diseases revealed patterns of structure−function relationships that may govern disease development and pathogenesis11,18,19. Additional investigation of deregulated kinase activities in cancer by pathway analysis using phospho-proteomics, analysis of gene amplifications/mutations and the generation of new kinase drugs has led to an improved understanding on how to use these KI drugs alone or in combination to obtain improved drug responses in patients1,14,20-22. The finding that protein kinases with a gain of function mutation appear to be either more sensitive (and sometimes resistant) to KIs compared to the wild type variant enabled the design of novel second generation KIs1,12,14,23-26. Protein kinases escape resistance either by mutating key residues in their catalytic domains or by reactivating pathways allowing bypassing the kinase target by overexpression of alternative kinases and/or other oncogenes27-34. Therefore, strategies have been deployed to override these various types of resistances including compounds capable of circumventing the target related drug resistance35-40. In this review, we will focus on the conformational bias and mode of binding of KIs because they determine their off-rates, residence times, selectivity and toxicity profile and ultimately disease modulation and efficacy.
Protein kinases act as molecular switches
Protein kinases can be viewed as molecular switches that amplify signals by passing them on to downstream targets via phosphorylation. Most eukaryotic protein kinases, with the exception of the atypical and the pseudo kinases, contain conserved catalytic residues instrumental for the transfer of the gammaphosphate from ATP to S, T and/or Y residues of target proteins. Regulatory domains are present either within or outside the kinase domain and help both to localise the kinase and to modulate its activity in response to various stimuli.
Typically, the kinase domain consists of 250–300 amino acids with 12 conserved subdomains that fold into a common catalytic core structure41. Many structures have been solved of protein kinases allowing mapping the subdomains41 onto the structure resulting in the visualisation of the glycine-rich loop (G-loop), catalytic loop, hinge as well as the activation loop (A-loop) (Figure 1). The kinase domain is a bi-lobal structure with the N-terminal lobe consisting mainly of beta-sheets and the C-terminal domain of alpha helices15,18,21,42-45. The two lobes are connected by the hinge region that lines the ATP-binding site that is targeted by the majority of small molecular weight KIs. The amino-terminal (N)-lobe of the catalytic domain contains the G-loop a stretch of residues that is crucial for ATP binding and phosphoryl transfer. The C-terminal lobe of the kinase domain is involved in substrate binding, and contains a conserved aspartic acid that is important for the catalytic activity of the enzyme15,18,21,42-45.

Protein kinases are turned on and off by very precise biological cues in which the helix C (the sole helical structure in the N-terminal lobe), the G-loop and the A-loop play important regulatory roles in the dynamic protein kinase domain. In particular, the A-loop with its helical sub-domain provides docking sites for protein/peptide substrates.
Conformation and binding modes
To fine-tune phosphorylation of substrates, most protein kinases adopt on- and off-states with corresponding mechanisms of down regulation and/or activation. A comprehensive structural, biophysical and biochemical information revealing the various transitions from the fully inactive to the fully activated state of a kinase is available only for a few protein kinases46,47. Not all protein kinases undergo this cycle of activation/deactivation. Some kinases, like CK2, are always in the on-state and only the location of the kinase determines its substrate selectivity48.
All active kinases share close commonalities for catalysis18 while the mechanisms of down regulation leading to the off-state as well as the conformation of the inactive kinase domains can be very diverse. Two textbook examples of an active conformation are revealed in the ternary complexes of the Insulin receptor (InsR), a receptor tyrosine kinase (RTK)(1ir3) and the cAMP-dependent serine/threonine-specific protein kinase (STK) PKA (1l3r) bound to nucleotide and substrate46,49. The latter structure is particularly instructive, as it features a transitions state mimetic corresponding to the ortho-phosphate during phosphate transfer. The two Mg2+-ions are positioned by conserved residues from the DFG (the very N-terminal portion of the A-loop) and catalytic loop and coordinate the ATP-phosphates. The G-loop clamps down on ATP, coordinating and shielding the gamma-phosphate from water. The A-loop is well ordered and forms a docking groove for the peptide substrate, poising the phospo-acceptor for transfer (Figure 1, above).
In this review, we define a conformation as active solely by the backbone conformation of the DFG loop and helix C similar to that seen in 1ir3, i.e. the DFG-motif forms the typical turnhairpin- turn conformation with the helix C in tight contact with the DFG (Figure 1, above). This definition of the active conformation captures the required elements of all active kinases, but may not be equivalent with activity in the biochemical sense. Depending on the kinase, additional regulatory elements outside of the kinase domain may be required for activity (e.g. cyclin binding, SH2-SH3 domains etc.). Unfortunately, the low degree of conservation of these regulatory elements, does not lend them to generalisation. By extension, conformations are defined as inactive if they do not meet the prerequisites of an active conformation and can be further qualified as helix C-out and/or DFG-out.

Conformation provides a common framework for understanding the activation of the kinase, disease causality, therapeutic modalities and resistance. The majority of kinase deregulation involves activation of kinases and can be understood in terms of the equilibrium between active and inactive conformation, which governs the fraction of the switch that is in the on-state. There are many ways by which kinase can be deregulated by pushing the equilibrium towards a constitutive active conformation although a comprehensive structural and biochemical elucidation of this process is only slowly emerging (Figure 2, above). Kinase activity can be increased by either over-expression of the kinase domain (gene amplification), chromosomal rearrange – ments (translocations, inversions) and activating mutations (missense, deletions, insertions)1,50,51. Activating mutations can either occur in the kinase domain itself, like K506E in FGF-R3, D816V in Kit, D842V in PDGFRα, and M918T in RET or located outside of the active kinase domain like the V617F in the pseudokinase domain of JAK2, juxta-membrane (JM) domain of Kit, PDGFR or Fms as well as in the extracellular binding domain like in Kit, RET and other RTKs, just to cite a few19,51,21. This view may be biased by the overrepresentation of receptor and nonreceptor TKs among the kinase disease targets, as their regulation often relies on a native inactive state. This is in stark contrast to other kinase families, like the AGC family of kinases which tend to have native active conformations and which are usually down regulated by additional regulatory elements. The bias towards the inactive state can be overcome by a multitude of factors which pulls the kinase towards its active conformation like mutations, insertions, deletions, dimerisation, nucleotide binding, KI binding (active conformation, normal koff), effector binding (cyclin, PIF pocket, SH3/SH2 etc.) as well as phosphorylation (Figure 2, page 44). In this view, the mode of action of the KI is crucial as different modes of action can have opposing effects on con for – mation, to the point that the activating effect of an inhibitor can overcome the inhibitory effect. Understanding kinase deregulation from a conformational perspective also accommodates the important aspects of downstream signalling via priming and docking, in addition to phosphorylation.
Classification of kinas conformations and binding modes

Excellent reviews have discussed the aspects of design principles, scaffolds, selectivity and particular binding modes40,53,54. A popular classification of the binding mode of KI is the distinction between type 1 and type 2 binders as first proposed by Li55. The type 2 KIs are defined as those targeting a DFG-out conformation in which a hydrophobic moiety fills the space vacated by the Phe (F) of the DFG motif and a crucial donor-acceptor motif interacts with the Asp (D) of DFG and/or catalytic Lys (K) and/or Glu (E) of helix C. Type 1 KIs are defined as inhibitors that occupy the ATP site but do not require a DFG-out conformation. This definition has been extended to the intermediate type ‘1.5’, essentially a type 2 KI with a smaller back pocket moiety binding to a DFG-in conformation56. Recently, several allosteric KIs have been structurally characterised as occupying a pocket outside of the ATP site2,57. Although the diversity of the binding sites reflects their excellent selectivity profiles, allosteric KIs are often jointly referred to as type 354,55,58. The binding sites of structurally characterised allosteric Kis are shown in Figure 3, above. The type 1 to type 3 definition is popular and has an appealing simplicity. However, the simplicity of this classification also limits its usefulness in discussing conformational aspects. In the daily practice, the added qualification of type 1 as ‘not necessarily’ binding to a DFG-out conformation is usually forgotten and superseded by a pharmacophore view, which classifies type 2 KIs based on the ligand structure. The pharmacophore definition of type 1 and 2 ignores conformational aspects and equates type 1 with the active, type 2 with the inactive DFG-out conformations. There are multiple examples of type 2 KIs in the pdb that bind to DFG-in conformations. In some cases, different protein units of the same crystal structure show DFG-in and DFG-out conformations, like the BRAF inhibitor PLX4720 (pdb 3c4c). Sometimes both conformations exist simultaneously as demonstrated by the four conformations of the type 1 KI SB203580 with p38α, which are once DFG-in (pdb 1a9u), once DFG-out (pdb 3gcp) and once in both (pdb 2ewa) (Figure 4). These structures emphasise the equilibrium nature of conformations that calls for a thermodynamic view. Furthermore, there are many types of inactive conformations and not all of them have DFG-out as their defining criterion. A more detailed classification of inactive kinase conformations would be needed to offer a meaningful differentiation. For lack of a better definition, we further qualify inactive conformations with helix C-in/out and DFG-in/out, where needed.

Similar confusion reigns around the term ATP-competition, a biochemical concept that is often falsely extrapolated to binding and structure. The term ATP-site binder is preferred and denotes a KI that binds to the ATP-binding pocket, overlapping with the ATP-binding. Although most type 1 ATP-site binders show ATP-competition, there are – admittedly puzzling – examples which do not59. The type 2 KIs are ATP-competitive as their target conformation is in equilibrium with an active, ATP-binding conformation (Figure 3). More importantly, lack of ATP-competition is not necessarily indicative of an allosteric or exo-site binding. The type 3 KIs like the MEK1/2 KI60 and ABL myristate pocket binder40,61 are truly non- ATP-competitive while the mixed-competition for the CHK1 inhibitors which according to structure also would classify as type 3, reminds us that competition in binding and functional inhibition are not identical57. Thus shifting from a ‘Ki’ (i.e. kinetic) to a ‘Kd’ (i.e. binding) approach may solve some of these discrepancies. As biochemical screening is the mainstay in the pharmaceutical industry, the recent trend towards profiling inhibitor activities in terms of binding (Ambit/DiscoverX, SPR and biophysical methods, cellular pull-down like Kinativ etc.) in conjunction with structure will further our knowledge for the binding mode of KI.
Conformational penalty and conformational bias

In theory, every conformation observed in a kinase is accessible in any other kinase at an energetic cost where the conformational penalty of a bound conformation is the difference to the minimum energy conformation (Figure 5, above). Conformational bias can be viewed as any event that is associated with a shift in conformational equilibrium, be it by binding, mutation or covalent modifications. We distinguish between the intrinsic con – formational penalty of the non-phosphorylated kinase domain that depends only on sequence, and extraneous conformational biases that lie outside of the kinase domain (e.g. JM, TM, SH2/SH3) or modify the kinase domain chemically or by binding (phosphorylation, coregulator binding, like cyclin). In principle, conformational equilibrium can be best investigated in solution by NMR, but due to high technical barriers (protein consumption, protein stability, structure assignment, etc.) only a few comprehensive studies have been reported thus far for protein kinases62.
Another way to estimate the contribution of conformational bias is by comparing various activating and inactivating mutants, different activation states, molecular ensembles as well as the related protein kinases. Based on an analysis of experimental data, Kufavera and Abagyan estimated a conformational energy difference of 0.7 to 4.0 kcal/mol for ABL, LCK and SRC relative to its phosphorylated state63 while the kinetic parameters for the pre-equilbrium between active and inactive conformation for imatinib binding to ABL by Shan et al. correspond to a conformational energy difference of 0.3 kcal/mol64.
Although a quantitative understanding of conformational penalty is still in its infancy, it helps to conceptualise phenomena like crossreactivity and the low ligand efficiency of slow binders. The type 2 or the lapatinib-like (‘type 1.5’) KIs typically require a higher molecular weight than type 1 KIs to achieve two-digit nano-molar potency. The assumption is that the intrinsic binding affinity of a potent type 1 is similar to a potent type 2 KI, but offset by the conformational penalty. If the interaction is not productive (low intrinsic ligand efficiency) and/or the conformational penalty too high, selectivity towards related kinases may increase like in the case of the higher affinity of imatinib for ABL compared to the closely related SRC. The identification of potent, structurally characterised type 2 KIs with inverted selectivity for SRC over ABL demonstrates that both conformational penalty and intrinsic ligand efficiency play into the observed potency65,66.
Conformation and binding kinetics
Binding kinetic has an important conformational component as binding to high energy conformations leads to slow binding (Figure 5A). Besides the simple binding (A), two additional scenarios can be distinguished. In the induced fit binding model (B), the conformational change that leads to a more productive binding takes place after ligand binding (I’). A more recent concept is that of conformational selection, in which the ligand binds to a low abundance, preformed bound conformation (E’), which is rapidly replenished from the equilibrium.
(A) simple binding E + I = EI
(B) induced fit binding E + I = EI = EI’
(C) conformational selection binding E + I = E’ + I = EI
Figure 5B shows a collection of bind – ing kinetics of KI reported in the literature67-69. ATP-site binders that bind to active conformations, where the bound conformation is energetically similar to that of the native conformation, typically have on-rates in the order of 106 M-1s-1. By contrast, ATP shows a very rapid binding and its on-rate is diffusion-limited. Many KIs that bind to inactive conformations have been characterised as having slower onrates in the order of 102 to 103 M-1s-1 (BIRB796 in p38α, sorafenib in BRAF, imatinib in ABL1, laptinib in EGFR etc.). The reason for the slower on rates is that the bound conformation carries an energetic penalty and is, therefore, poorly populated in equilibrium, rendering the global conformational change rate-limiting for binding. The case for conformational selection has been made convincingly for the pHdependent binding of imatinib to ABL where the DFG-out conformation is first generated in a rate-limiting step (kconf ~ 100 s-1), followed by ligand binding with an on-rate similar to that observed for active conformation binders64. These results also indicates that an induced fit model is difficult to reconcile with the mutually exclusive structural requirements of type 2 KI binding and the DFG-out transition70.
Tummino et al. have argued against con – formational selection for type 2 KIs, even though an induced fit model is difficult to reconcile with the mutually exclusive structural requirements of type 2 KI binding and the DFG-out transition83. A corollary of the slow binding is a slow off-rate leading to a prolonged residence time on the target kinase that can be important for pharmacological selectivity and efficacy. The importance of the residence time for the in vivo efficacy has been discussed in recent reviews70-72.
Conformation and selectivity
What degree of selectivity a KI should ideally have is, to date, a controversial issue73-78. Selectivity is an important key parameter of medicinal chemistry efforts and many projects prefer to err on the side of selectivity, rather than assessing the balance between toxicity and efficacy on a compound per compound basis. A look at clinically advanced KIs reminds us that a promiscuous selectivity profile as that of sunitinib can be tolerated in oncological settings, albeit with some major side effects. On the other hand, even some of the most selective KI for mTOR, MEK and p38 can have serious on-target related toxicologies73-78. With the exception of the highly selective lapatinib, the other marketed KIs derive at least in part their efficacy from poly-pharmacy73-78. In any case, for pharmacologic target validation as well as chronic administration of KIs in non-oncological indications, however, a reasonable selectivity is a prerequisite which is being achieved79.
Among various providers that offer kinase selectivity panel, the so-called Ambit-panel (now part of DiscoverX) has the broadest kinome coverage and has established itself as one of the industries’ gold standard for assessing selectivity74,76-78. For the assessment of selectivity, caveats regarding assay formats, correlation to cellular selectivity, activation state of the recombinant kinase and the usually poor physicochemical properties of most KIs, require a cautious interpretation of flags from these panel data. The most important contribution of kinase panels is probably the cross-fertilisation between protein kinase projects. Unfortunately, there is a perception that there is a trend towards higher selectivity going from type 1 to type 2, while it is undisputed that type 3 KIs display the highest degree of selectivity. The implication that type 2 KI are generally more selective than type 1 lacks quantitative arguments. In fact there are examples of exquisite selective KIs known for all types. Selectivity rests on particular features of a particular protein kinase, regardless of the binding mode of the KI. As DFG-out con – formations are accessible only in about 10 per cent of the kinome (judging by the abundance in the pdb), the type 2 KI always have a head start regarding selectivity because only the so called DFG-out competent kinases are targeted in the kinome. In the final stages of compound optimisation, selectivity is typically already quite advanced and the objective is rather to dial out a few remaining ‘unwanted’ protein kinase targets in addition to optimise the pharmacologic properties. In this context, the extended pharmacophore of the type 2 KIs may be more detrimental as it involves interaction with many conserved residues. The selectivity profile of type 2 KIs is inherent to a scaffold from the very start and difficult to fine-tune merely by changing the substitution pattern, as the targets tend to cluster in families. Type 1 KIs on the other hand interact with a higher proportion of nonconserved residues and, provided selectivity elements can be identified, even promiscuous scaffolds can be made selective by the addition of a few judiciously chosen substituents. In contrast to type 1 and 2, the truly allosteric type 3 KI have thus far lived up to the expectations regarding selectivity, as demonstrated by the few examples of potent type 3 KIs disclosed so far40,61. The high level of selectivity reflects the unique binding sites and is off-set by the difficulty of obtaining and optimising chemical matter (Figure 3)57,80. So far, there are only sparse hints that allosteric sites can be generalised and chemical matter transferred to other kinases81.
Sequence determines selectivity not only via stabilising a conformation, but also on an interaction-base by determining the side-chains that interact with the ligand. A priori, every conformation should be accessible by every kinase with the sequence determining its stability relative to its minimum energy or the same conformation in other kinases or mutants thereof.
The most rational way to obtain selectivity is interaction-based, by targeting poorly conserved residues in particular residues flanking the hinge, like the gatekeeper, which controls access to the back pocket, and for example the gatekeeper+2, which closes off a hydrophobic rim of the hinge. An example for the importance of the gatekeeper is the wellknown SB203580 which is selective for p38α over p38γ and whose distal aryl ring is accommodated in the back-pocket, bypassing the gatekeeper Thr (pdb 1a9u). The closely related p38γ/p38δ-isoforms carry a bulky MET gatekeeper which blocks access to the back pocket and makes them insensitive to inhibition by SB203580. Similarly, the closely related kinases ERK and JNK3 which feature a MET gatekeeper can be made sensitive to SB203580 analogs by mutation of the gatekeeper from MET to Thr82,83.
Another instructive example in which several sequence features are exploited, is the PLK inhibitor BI-253684 where in the pyrimidopyridone scaffold, an ethyl moiety of BI-2536 targets a space vacated by a Cys in the G-loop (normally Val) and its o-methoxy-aniline targets the smaller gatekeeper+2 Leu (normally Phe/Tyr, pdb 2rku). Thus, two small substituents convert a rather promiscuous scaffold into a highly selective inhibitor.
Interaction-based selectivity also exists for type 2 KI like the Amgen’s MET KI, which targets a unique Gly-‘hole’ in helix C (pdb 3f82)85,86. In all of the above cases, the target conformations in the anti-targets would be energetically similar and therefore accessible, but the interactions would be non- or counter-productive due to for example steric clash.
Although typically discovered by seren – dipity rather than rational design, the interplay of sequence and conformational penalty can lead to exceptional selectivity. The balance between sequence and conformational contributions comes in different flavours. In one extreme, optimisation of the compound leads from an active to an inactive, high energy conformation of the kinase such that the additional interactions make up for the conformational penalty, but only on the target kinase. Examples are lapatinib vs. the relatively unselective erlotinib (helix C-out) and imatinib in ABL vs. SRC (DFG-out)87,88.
Another example that is both interactionbased as well as conformation-driven is the MET kinase whose native inactive conformation offers the potential for a unique, crucial stacking interaction with an A-loop Tyr (pdb 3ccn)89. Both the shape of the resulting pocket as well as the subtle stacking interactions are key for highly selective inhibitors. Here, several unique sequence features play together to stabilise an otherwise high-energy conformation.
Buriedness of the ATP-site can influence selectivity by making similar interactions more productive. A factor that is often involved is the ability of a kinase’s G-loop to partially collapse onto the ligand, thus creating a more buried, less solvent-exposed cavity with high intrinsic ligand efficiency. This can lead to a low level of selectivity despite of highly similar binding modes and similar conformations. Examples of kinases that show high intrinsic ligand efficiency include ABL, Aurora and GSK3β. Whether this apparent selectivity translates into cellular activity or is merely caused by a very sensitive biochemical assay needs to be checked case by case.
Lastly, there are cases of excellent selectivity that are not easily explained by any single factor and arise from a multitude of subtle interactions. An example is the exquisite selectivity of Pfizer’s JAK inhibitor CP-690,550, which, again, builds upon a box-standard, pan-KI pyrrolo-pyrimidine scaffold79,90. Although it targets an active conformation, the combination of shape, a unique G-loop conformation and subtle interactions between backbone carbonyls and the inhibitors nitrile group lead to a surprising level of selectivity (pdb 3fup)90.
Conformational penalty and resistance
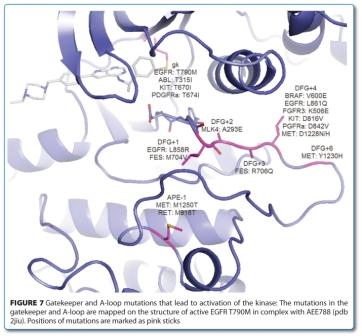
Resistance to KI can emerge in several ways during targeted therapy, raising the issue of an endless chase of resistance alleles, with ever more specific inhibitors28,40,61,91. Apart from the usual mechanisms of resistance that tend to decrease the level of the inhibitors in cells, the resistance mechanisms to KI act in large part to restore the activity of the original ‘canceraddicting’ pathway. This can occur either by conformational changes in the kinase domain or by reactivating the pathway downstream and/ or parallel to the targeted kinase31. In this case, inhibitors targeting inactive conformations are generally more prone to resistance mutations as a single mutation can act both by destroying crucial interactions and/or destabilising the target conformation. Two frequent types of activating mutation, gatekeeper and A-loop mutations stand out and serve to illustrate the link between mutation and conformation. While the gatekeeper mutations is well conserved92 the A-loop mutations are diverse93 (Figure 7). Both types of mutations activate by shifting the equilibrium to the active state and thereby increasing the (biochemical) activity of the protein kinase92. In addition to blocking access to the back pocket (which is detrimental to ligand binding), mutation of a small to a large hydrophobic gatekeeper also stabilises the active conformation, presumably by stabilisation of the so called hydrohobic spine – a network of hydrophobic interactions in the kinase domain – that promote the assembly of the active kinase conformation18,94.
ABL inhibitors capable of both directly suppressing such mutations and more potently inhibiting the wild-type kinase have sub – stantially improved the molecular response rates61,95,96. Although clinical remission is achieved with these second generation inhibitors like dasatinib and nilotinib, the gatekeeper T315I mutation has still remained elusive95,96. The ABLT315I drug-resistant mutant biases the kinase towards an active conformation thereby blocking access to the back-pocket and eliminating a crucial hydrogen bond to the T315 hydroxyl group12,18. Similarly, activating mutation of the gatekeeper causes resistance of KI to imaitinib (D816V) in systemic mastocytoma51 and of EGFR to lapatinib/ gefitinib (T760M)13,28. In case of the EGFR, activating and/or drug resistant mutations can lead to an increased affinity for ATP that loosens the binding to gefitinib97,98.
Conformational bias can also contribute to efficacy if the inhibitor preferentially binds to an active conformation like in the case of the activating mutation of EGFR23. If the toxicity has an on-target component, the preferential targeting of the mutated activated kinase in diseased tissue can also improve the therapeutic index, as in the case of BRAF V600E and EGFR which can be extended obviously to the other activating mutations like those in type IIIs RTPKs93. Lastly, unlike in the previous examples that only involve the catalytic domain, kinases are often regulated by additional domains. The logic of conformational bias also applies in this case and the binding of allosteric effectors can shift equilibrium to effectively lock the kinase in a given conformation, which opens the possibility to activate or inhibit the kinase. The recently discovered allosteric myrpocket inhibitors (Figure 3, page 46) assembles the inactive conformation of Abl or BcrAbl by promoting the docking of the SH3 and SH2 domains onto the Abl kinase domain, which seems to also work for the T315I mutant40,61. Combination of inhibitors that bind to the myrpocket leading to the assembly of the inactive conformation of BcrABL and ATP-site inhibitors may become clinically useful in overcoming and suppressing the emergence of the major imatinib-, nilotinib- and dasatinib- resistant T315I mutation40,61. Ligand binding to the myristate pocket mimics the myristoylation of Gly2 of ABL that acts as a conformational bias to lock the kinase in its inactive state. Accordingly, G2A mutation already leads to partial activation by weakening the assembly of the inactive state92.
Dimerisation of kinases, in particular the ligand induce RTPKs, is another common mechanism for the activation of kinases99. Asymmetric dimerisation presumably acts as another conformational bias that pushes down helix C to a more active conformation in analogy to activation of CDKs by cyclin binding99-101.
Another interesting and completely diverse way to generate resistance to KI is exemplified by the B-RAF where a gain-of-function mutation in the B-RAF gene is responsible in about, and its sustained expression is required for tumour-cell proliferation and survival 50 per cent of human melanomas102. Vemurafenib (PLX4032), a KI which target the most prevalent oncogenic B-RAF V600E variant, has been recently approved against metastatic melanoma but unfortunately, most patients acquire resistance to vemurafenib within a year of treatment103-105. Most modes of resistance discovered so far involve circum vention of this mutant B-RAF leading to reactivation of the ERK pathway via alternative entry points106-108. Reactivation can involve either KI-induced heterodimerisation with c-RAF or via enhanced dimerisation of the kinase domain of a mutant (truncated) form of B-RAF-V600E that cannot be inhibited by vemurafenib109 indicating that dimerisation normally activates the wild-type protein on association with signal-activated RAS110. Paradoxical activation of BRAF by inhibitors occurs by KI-induced hetero-dimerisation of cRAF/BRAF that leads to pathway activation111,112. How inhibitor binding achieves transactivation merely by binding is unclear. An explanation could be that the KI acts as a transitory conformational bias that pulls cRAF/BRAF towards a more active conformation with increased capacity to activate cRAF via dimerisation. Interestingly, the inactive conformation binder AZ-628, a type 2 KI, does not lead to activation112.
Thus, it appears that kinases can escape drug resistance by many diverse mechanisms. Only a comprehensive combination of inhibitors which take care of the resistance in the target kinase as well as of the various modes of the reactivation of the pathway(s) will be required to combat the emerging resistance in targeted therapies.
Conclusions
Protein kinases are molecular switches that adopt on- and off-states with corresponding mechanisms of down regulation and/or activation. Understanding the conformational changes that leads from one to the other state of activation will allow for a better design of KIs. In addition, understanding conformations will also provide a common framework for understanding the activation of the kinase, disease causality, therapeutic modalities and resistance. Activation and deregulation of protein kinases occur by pushing the equilibrium towards a constitutive active conformation which is very similar in all protein kinases and in its essence can be defined by the DFG motif forming the typical turn-hairpin-turn conformation in tight contact with helix C. Activation (deregulation) of protein kinases can be induced by mutations within or outside of the kinase domain as well as by regulatory elements. In contrast, conformations of inactive kinase domains are very diverse and can be qualified as helix C-out and/or DFG-out. This has also provided the basis for the popular, albeit limited, classification of the binding modes of KIs into type 1, type 2 and type 3. One weakness of this classification is the fact that it equates type 1 with the active, type 2 with the inactive DFG-out conformation.
The mode of action of KIs is linked to their binding mode, i.e. target conformation which has implications on binding kinetics and selectivity. Using biophysical methods in conjunction with structures will further our knowledge of the binding mode of KIs. Ideally, the shift in conformational equilibrium, be it by binding, mutation or covalent modifications, would be studied in solution using NMR. This allows quantifying and better understanding conformational penalty and conceptualising selectivity and slow binding. This thinking has led to the concept of conformational selection where the KI binds to a low abundance preformed target conformation. The slow on rate is a consequence of the conformational penalty of the target conformation that is, therefore, poorly populated in equilibrium, rendering the global conformational change rate-limiting for binding. The selectivity of KIs often stems from binding to high energy conformations, whereas the most rational way to obtain selectivity is interaction-based. The most striking examples of selectivity come from targeting unique allosteric sites. On the other hand, targeting an active conformation can also result in excellent selectivity that are not easily explained by any single factor and arise from a multitude of subtle interactions. Mutations act as another conformational bias and depending on the mode of action can lead to resistance or contribute to the efficacy of KIs.
In summary, conformation provides a common framework for understanding the activation of the kinase, disease causality, therapeutic modalities and resistance.
About the authors
Originally an organic chemist, Henrik Möbitz obtained his PhD in bio chemistry at the Albert- Ludwigs-Universitat Freiburg in 2003. After a postdoctoral stay in computational chemistry at the UC Santa Barbara with Tom Bruice, he joined the Computer-Aided Drug Design group of Novartis in 2005. Henrik Möbitz has a keen interest in kinase biology and medicinal chemistry. He enjoys the global collaborations, the data rich environment and working at the interface between medicinal chemistry, modeling, structural biology, biophysics and in vitro biology, all the way to pharmacology.
Doriano Fabbro received his PhD in cell biology and biochemistry at the Biocenter Basel, Switzerland, for the studies on the mechanisms of activation of PKC. In 1991, he joined the Oncology Research group of Ciba-Geigy Basel where he contributed to the discovery of Midostaurin as well as Glivec. Following the merger of Ciba-Geigy with Sandoz to form Novartis in 1996, he was promoted to Executive Director in Oncology Research directing drug discovery efforts focusing on ATP-dependent enzymes (protein kinases and ATPases) as well as other small molecular weight approaches. In 2006, he was instrumental for the establishment of NIBR Expertise Kinase Platform where he serves as Head of the Kinase Biology. Dr. Fabbro is a member of various professional societies and several journal editorial boards, publications and numerous patents in the area of protein kinases regulation, structure, screening and drug discovery. He has contributed to the discovery and development of various protein kinase inhibitors for the treatment of cancer including inhibitors. Among the compounds that reached full development/registration are RAD001 (Everolimus, Afinitor, launched), STI571 (Imatinib, Glivec, launched) and AMN107 (Nilotinib, Tasigna, launched) and PKC412 (Midostaurin, Ph III).
References
1. P. Blume-Jensen, T. Hunter, Oncogenic kinase signalling, Nature 411 (2001) 355-365
2. C.-J.S. Fabbro D, Möbitz H, Martiny-Baron G., Targeting cancer with small-molecular-weight kinase inhibitors, Methods Mol Biol 795 (2012) 1-34
3. C. Garcia-Echeverria, Protein and lipid kinase inhibitors as targeted anticancer agents of the Ras/Raf/MEK and PI3K/PKB pathways, Purinergic Signal 5 (2009) 117-125
4. L.K. Chico, L.J. Van Eldik, D.M. Watterson, Targeting protein kinases in central nervous system disorders, Nat Rev Drug Discov 8 (2009) 892-909
5. C. Garcia-Echeverria, W.R. Sellers, Drug discovery approaches targeting the PI3K/Akt pathway in cancer, Oncogene 27 (2008) 5511-5526
6. S.M. Maira, C. Voliva, C. Garcia-Echeverria, Class IA phosphatidylinositol 3-kinase: from their biologic implication in human cancers to drug discovery, Expert Opin Ther Targets 12 (2008) 223-238
7. T.L. Yuan, L.C. Cantley, PI3K pathway alterations in cancer: variations on a theme, Oncogene 27 (2008) 5497-5510
8. M. Malumbres, M. Barbacid, Cell cycle, CDKs and cancer: a changing paradigm, Nat Rev Cancer 9 (2009) 153-166
9. J. Luo, N.L. Solimini, S.J. Elledge, Principles of cancer therapy: oncogene and non-oncogene addiction, Cell 136 (2009) 823-837
10. I.B. Weinstein, Cancer. Addiction to oncogenes–the Achilles heal of cancer, Science 297 (2002) 63-64
11. W.R. Sellers, A blueprint for advancing genetics-based cancer therapy, Cell 147 (2011) 26-31
12. D. Fabbro, Fendrich, G., Guez V, Meyer T, Furet P, Mestan P, Griffin JD, Manley PW, Cowan-JacobSW, Targeted therapy with imatinib: An exception or a rule? , Handbook of Experimental Pharmacology, Inhibitors of Protein Kinases and Protein Phosphates 167 (2005) 361-389
13. W. Pao, V.A. Miller, K.A. Politi, G.J. Riely, R. Somwar, M.F. Zakowski, M.G. Kris, H. Varmus, Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain, PLoS Med 2 (2005) e73
14. C. Sawyers, Targeted cancer therapy, Nature 432 (2004) 294-297
15. A. Bardelli, D.W. Parsons, N. Silliman, J. Ptak, S. Szabo, S. Saha, S. Markowitz, J.K. Willson, G. Parmigiani, K.W. Kinzler, B. Vogelstein, V.E. Velculescu, Mutational analysis of the tyrosine kinome in colorectal cancers, Science 300 (2003) 949
16. C. Greenman, P. Stephens, R. Smith, G.L. Dalgliesh, C. Hunter, G. Bignell, H. Davies, J. Teague, A. Butler, C. Stevens, S. Edkins, S. O’Meara, I. Vastrik, E.E. Schmidt, T. Avis, S. Barthorpe, G. Bhamra, G. Buck, B. Choudhury, J. Clements, J. Cole, E. Dicks, S. Forbes, K. Gray, K. Halliday, R. Harrison, K. Hills, J. Hinton, A. Jenkinson, D. Jones, A. Menzies, T. Mironenko, J. Perry, K. Raine, D. Richardson, R. Shepherd, A. Small, C. Tofts, J. Varian, T. Webb, S. West, S. Widaa, A. Yates, D.P. Cahill, D.N. Louis, P. Goldstraw, A.G. Nicholson, F. Brasseur, L. Looijenga, B.L. Weber, Y.E. Chiew, A. DeFazio, M.F. Greaves, A.R. Green, P. Campbell, E. Birney, D.F. Easton, G. Chenevix-Trench, M.H. Tan, S.K. Khoo, B.T. Teh, S.T. Yuen, S.Y. Leung, R. Wooster, P.A. Futreal, M.R. Stratton, Patterns of somatic mutation in human cancer genomes, Nature 446 (2007) 153-158
17. R.K. Thomas, A.C. Baker, R.M. Debiasi, W. Winckler, T. Laframboise, W.M. Lin, M. Wang, W. Feng, T. Zander, L. MacConaill, J.C. Lee, R. Nicoletti, C. Hatton, M. Goyette, L. Girard, K. Majmudar, L. Ziaugra, K.K. Wong, S. Gabriel, R. Beroukhim, M. Peyton, J. Barretina, A. Dutt, C. Emery, H. Greulich, K. Shah, H. Sasaki, A. Gazdar, J. Minna, S.A. Armstrong, I.K. Mellinghoff, F.S. Hodi, G. Dranoff, P.S. Mischel, T.F. Cloughesy, S.F. Nelson, L.M. Liau, K. Mertz, M.A. Rubin, H. Moch, M. Loda, W. Catalona, J. Fletcher, S. Signoretti, F. Kaye, K.C. Anderson, G.D. Demetri, R. Dummer, S. Wagner, M. Herlyn, W.R. Sellers, M. Meyerson, L.A. Garraway, High-throughput oncogene mutation profiling in human cancer, Nat Genet 39 (2007) 347-351
18. S.S. Taylor, A.P. Kornev, Protein kinases: evolution of dynamic regulatory proteins, Trends Biochem Sci 36 (2011) 65-77
19. P. Lahiry, A. Torkamani, N.J. Schork, R.A. Hegele, Kinase mutations in human disease: interpreting genotypephenotype relationships, Nat Rev Genet 11 (2010) 60-74
20. P. Cohen, Protein kinases–the major drug targets of the twenty-first century?, Nat Rev Drug Discov 1 (2002) 309- 315
21. M. Vieth, R.E. Higgs, D.H. Robertson, M. Shapiro, E.A. Gragg, H. Hemmerle, Kinomics-structural biology and chemogenomics of kinase inhibitors and targets, Biochim Biophys Acta 1697 (2004) 243-257
22. A. Wolf-Yadlin, N. Kumar, Y. Zhang, S. Hautaniemi, M. Zaman, H.D. Kim, V. Grantcharova, D.A. Lauffenburger, F.M. White, Effects of HER2 overexpression on cell signaling networks governing proliferation and migration, Mol Syst Biol 2 (2006) 54
23. J.G. Paez, P.A. Janne, J.C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F.J. Kaye, N. Lindeman, T.J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M.J. Eck, W.R. Sellers, B.E. Johnson, M. Meyerson, EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy, Science 304 (2004) 1497-1500
24. W. Zhou, D. Ercan, L. Chen, C.H. Yun, D. Li, M. Capelletti, A.B. Cortot, L. Chirieac, R.E. Iacob, R. Padera, J.R. Engen, K.K. Wong, M.J. Eck, N.S. Gray, P.A. Janne, Novel mutantselective EGFR kinase inhibitors against EGFR T790M, Nature 462 (2009) 1070-1074
25. M.C. Heinrich, C.L. Corless, G.D. Demetri, C.D. Blanke, M. von Mehren, H. Joensuu, L.S. McGreevey, C.J. Chen, A.D. Van den Abbeele, B.J. Druker, B. Kiese, B. Eisenberg, P.J. Roberts, S. Singer, C.D. Fletcher, S. Silberman, S. Dimitrijevic, J.A. Fletcher, Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor, J Clin Oncol 21 (2003) 4342-4349
26. T.J. Lynch, D.W. Bell, R. Sordella, S. Gurubhagavatula, R.A. Okimoto, B.W. Brannigan, P.L. Harris, S.M. Haserlat, J.G. Supko, F.G. Haluska, D.N. Louis, D.C. Christiani, J. Settleman, D.A. Haber, Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib, N Engl J Med 350 (2004) 2129-2139
27. M.E. Gorre, M. Mohammed, K. Ellwood, N. Hsu, R. Paquette, P.N. Rao, C.L. Sawyers, Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification, Science 293 (2001) 876-880
28. S. Kobayashi, T.J. Boggon, T. Dayaram, P.A. Janne, O. Kocher, M. Meyerson, B.E. Johnson, M.J. Eck, D.G. Tenen, B. Halmos, EGFR mutation and resistance of non-smallcell lung cancer to gefitinib, N Engl J Med 352 (2005) 786-792
29. T. Hunter, Signaling–2000 and beyond, Cell 100 (2000) 113-127
30. J.J. Ventura, A.R. Nebreda, Protein kinases and phosphatases as therapeutic targets in cancer, Clin Transl Oncol 8 (2006) 153-160
31. J.A. Engelman, K. Zejnullahu, T. Mitsudomi, Y. Song, C. Hyland, J.O. Park, N. Lindeman, C.M. Gale, X. Zhao, J. Christensen, T. Kosaka, A.J. Holmes, A.M. Rogers, F. Cappuzzo, T. Mok, C. Lee, B.E. Johnson, L.C. Cantley, P.A. Janne, MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling, Science 316 (2007) 1039-1043
32. T. Takano, Y. Ohe, H. Sakamoto, K. Tsuta, Y. Matsuno, U. Tateishi, S. Yamamoto, H. Nokihara, N. Yamamoto, I. Sekine, H. Kunitoh, T. Shibata, T. Sakiyama, T. Yoshida, T. Tamura, Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-smallcell lung cancer, J Clin Oncol 23 (2005) 6829-6837
33. S. Ali, S. Ali, Role of c-kit/SCF in cause and treatment of gastrointestinal stromal tumors (GIST), Gene 401 (2007) 38-45
34. S. Chandarlapaty, A. Sawai, M. Scaltriti, V. Rodrik- Outmezguine, O. Grbovic-Huezo, V. Serra, P.K. Majumder, J. Baselga, N. Rosen, AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity, Cancer Cell 19 (2011) 58-71
35. J.A. Engelman, L. Chen, X. Tan, K. Crosby, A.R. Guimaraes, R. Upadhyay, M. Maira, K. McNamara, S.A. Perera, Y. Song, L.R. Chirieac, R. Kaur, A. Lightbown, J. Simendinger, T. Li, R.F. Padera, C. Garcia-Echeverria, R. Weissleder, U. Mahmood, L.C. Cantley, K.K. Wong, Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers, Nat Med 14 (2008) 1351-1356
36. E. Weisberg, P.W. Manley, W. Breitenstein, J. Bruggen, S.W. Cowan-Jacob, A. Ray, B. Huntly, D. Fabbro, G. Fendrich, E. Hall-Meyers, A.L. Kung, J. Mestan, G.Q. Daley, L. Callahan, L. Catley, C. Cavazza, M. Azam, D. Neuberg, R.D. Wright, D.G. Gilliland, J.D. Griffin, Characterization of AMN107, a selective inhibitor of native and mutant Bcr- Abl, Cancer Cell 7 (2005) 129-141
37. A. Quintas-Cardama, H. Kantarjian, J. Cortes, Flying under the radar: the new wave of BCR-ABL inhibitors, Nat Rev Drug Discov 6 (2007) 834-848
38. F.J. Adrian, Q. Ding, T. Sim, A. Velentza, C. Sloan, Y. Liu, G. Zhang, W. Hur, S. Ding, P. Manley, J. Mestan, D. Fabbro, N.S. Gray, Allosteric inhibitors of Bcr-abl-dependent cell proliferation, Nat Chem Biol 2 (2006) 95-102
39. L.J. Lombardo, F.Y. Lee, P. Chen, D. Norris, J.C. Barrish, K. Behnia, S. Castaneda, L.A. Cornelius, J. Das, A.M. Doweyko, C. Fairchild, J.T. Hunt, I. Inigo, K. Johnston, A. Kamath, D. Kan, H. Klei, P. Marathe, S. Pang, R. Peterson, S. Pitt, G.L. Schieven, R.J. Schmidt, J. Tokarski, M.L. Wen, J. Wityak, R.M. Borzilleri, Discovery of N-(2-chloro-6- methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1- yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxami de (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays, J Med Chem 47 (2004) 6658-6661
40. J. Zhang, Adrian, F.J., Jahnke, W., Cowan-Jacob, S.W., Li,A.G., Iacob, R.E., Sim, T.,Powers, J. , Dierks, C. , Sun, F., Guo, G.R., Ding, Q., Okram, B. , Choi, Y.,Wojciechowski, A., Deng, X., Liu, G., Fendrich, G., Strauss, A., Vajpai, N., Grzesiek, S., Tuntland, T., Liu, Y., Bursulaya, B., Azam, M., Manley, P.W., Engen, J.R., Daley, G.Q., Warmuth, M., Gray, N.S., Targeting wild-type and T315I Bcr-Abl by combining allosteric with ATP-site inhibitors, Nature Chem. Biol. in press (2009)
41. S.K. Hanks, T. Hunter, Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification, Faseb J 9 (1995) 576-596
42. M. Vieth, J.J. Sutherland, D.H. Robertson, R.M. Campbell, Kinomics: characterizing the therapeutically validated kinase space, Drug Discov Today 10 (2005) 839-846
43. A. Levitzki, Protein kinase inhibitors as a therapeutic modality, Acc Chem Res 36 (2003) 462-469
44. D. Fabbro, C. Garcia-Echeverria, Targeting protein kinases in cancer therapy, Curr Opin Drug Discov Devel 5 (2002) 701-712
45. S.W. Cowan-Jacob, Structural biology of protein tyrosine kinases, Cell Mol Life Sci 63 (2006) 2608-2625
46. S.R. Hubbard, Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog, Embo J 16 (1997) 5572-5581
47. D.O. Morgan, The dynamics of cyclin dependent kinase structure, Curr Opin Cell Biol 8 (1996) 767-772
48. K. Niefind, J. Raaf, O.G. Issinger, Protein kinase CK2 in health and disease: Protein kinase CK2: from structures to insights, Cell Mol Life Sci 66 (2009) 1800-1816
49. Madhusudan, P. Akamine, N.H. Xuong, S.S. Taylor, Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase, Nat Struct Biol 9 (2002) 273-277
50. D. Fabbro, S.W. Cowan-Jacob, H. Mobitz, G. Martiny- Baron, Targeting cancer with small-molecular-weight kinase inhibitors, Methods Mol Biol 795 (2011) 1-34
51. J.A. Fletcher, B.P. Rubin, KIT mutations in GIST, Curr Opin Genet Dev 17 (2007) 3-7
52. S. Manie, M. Santoro, A. Fusco, M. Billaud, The RET receptor: function in development and dysfunction in congenital malformation, Trends Genet 17 (2001) 580-589
53. P. Traxler, G. Bold, E. Buchdunger, G. Caravatti, P. Furet, P. Manley, T. O’Reilly, J. Wood, J. Zimmermann, Tyrosine kinase inhibitors: from rational design to clinical trials, Med Res Rev 21 (2001) 499-512
54. J. Zhang, Yang, P. L., Gray, N. S., Targeting cancer with small molecule kinase inhibitors, Nat Rev Cancer 9 (2009) 28-39
55. B. Li, Y. Liu, T. Uno, N. Gray, Creating chemical diversity to target protein kinases, Comb Chem High Throughput Screen 7 (2004) 453-472
56. F. Zuccotto, E. Ardini, E. Casale, M. Angiolini, Through the “gatekeeper door”: exploiting the active kinase conformation, J Med Chem 53 (2010) 2681-2694
57. D. Vanderpool, T.O. Johnson, C. Ping, S. Bergqvist, G. Alton, S. Phonephaly, E. Rui, C. Luo, Y.L. Deng, S. Grant, T. Quenzer, S. Margosiak, J. Register, E. Brown, J. Ermolieff, Characterization of the CHK1 allosteric inhibitor binding site, Biochemistry 48 (2009) 9823-9830
58. Y. Liu, Gray, N. S., Rational design of inhibitors that bind to inactive kinase conformations, Nat Chem Biol 2 (2006) 358-364
59. Y. Blat, Non-competitive inhibition by active site binders, Chem Biol Drug Des 75 (2010) 535-540
60. J.F. Ohren, H. Chen, A. Pavlovsky, C. Whitehead, E. Zhang, P. Kuffa, C. Yan, P. McConnell, C. Spessard, C. Banotai, W.T. Mueller, A. Delaney, C. Omer, J. Sebolt-Leopold, D.T. Dudley, I.K. Leung, C. Flamme, J. Warmus, M. Kaufman, S. Barrett, H. Tecle, C.A. Hasemann, Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition, Nat Struct Mol Biol 11 (2004) 1192-1197
61. D. Fabbro, Manley, P.W., Jahnke, W., Liebetanz, J., Szyttenholm, A., Fendrich, G., Strauss, A., Zhang, J., Gray, N.S., Adrian, F., Warmuth, M. , Pelle, X., Grotzfeld, R., Berst, F., Marzinzik, A., Furet, P., Cowan-Jacob, S.W., Mestan, J. , Inhibitors of the Abl kinase directed at either the ATP- or myristate-binding site Biochem Biophys Acta in press. (2010)
62. N. Vajpai, A. Strauss, G. Fendrich, S.W. Cowan-Jacob, P.W. Manley, S. Grzesiek, W. Jahnke, Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib, J Biol Chem 283 (2008) 18292-18302
63. I. Kufareva, R. Abagyan, Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states, J Med Chem 51 (2008) 7921-7932
64. Y. Shan, M.A. Seeliger, M.P. Eastwood, F. Frank, H. Xu, M.O. Jensen, R.O. Dror, J. Kuriyan, D.E. Shaw, A conserved protonation-dependent switch controls drug binding in the Abl kinase, Proc Natl Acad Sci U S A 106 (2009) 139-144
65. A.C. Dar, M.S. Lopez, K.M. Shokat, Small molecule recognition of c-Src via the Imatinib-binding conformation, Chem Biol 15 (2008) 1015-1022
66. M.A. Seeliger, P. Ranjitkar, C. Kasap, Y. Shan, D.E. Shaw, N.P. Shah, J. Kuriyan, D.J. Maly, Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations, Cancer Res 69 (2009) 2384-2392
67. D.R. Anderson, M.J. Meyers, R.G. Kurumbail, N. Caspers, G.I. Poda, S.A. Long, B.S. Pierce, M.W. Mahoney, R.J. Mourey, Benzothiophene inhibitors of MK2. Part 1: structure-activity relationships, assessments of selectivity and cellular potency, Bioorg Med Chem Lett 19 (2009) 4878-4881
68. D.R. Anderson, M.J. Meyers, R.G. Kurumbail, N. Caspers, G.I. Poda, S.A. Long, B.S. Pierce, M.W. Mahoney, R.J. Mourey, M.D. Parikh, Benzothiophene inhibitors of MK2. Part 2: improvements in kinase selectivity and cell potency, Bioorg Med Chem Lett 19 (2009) 4882-4884
69. D. Casper, M. Bukhtiyarova, E.B. Springman, A Biacore biosensor method for detailed kinetic binding analysis of small molecule inhibitors of p38alpha mitogenactivated protein kinase, Anal Biochem 325 (2004) 126-136
70. P.J. Tummino, R.A. Copeland, Residence time of receptor-ligand complexes and its effect on biological function, Biochemistry 47 (2008) 5481-5492
71. D.C. Swinney, The role of binding kinetics in therapeutically useful drug action, Curr Opin Drug Discov Devel 12 (2009) 31-39
72. R.A. Copeland, D.L. Pompliano, T.D. Meek, Drug-target residence time and its implications for lead optimization, Nat Rev Drug Discov 5 (2006) 730-739.
73. R. Morphy, Z. Rankovic, Fragments, network biology and designing multiple ligands, Drug Discov Today 12 (2007) 156-160
74. M.W. Karaman, S. Herrgard, D.K. Treiber, P. Gallant, C.E. Atteridge, B.T. Campbell, K.W. Chan, P. Ciceri, M.I. Davis, P.T. Edeen, R. Faraoni, M. Floyd, J.P. Hunt, D.J. Lockhart, Z.V. Milanov, M.J. Morrison, G. Pallares, H.K. Patel, S. Pritchard, L.M. Wodicka, P.P. Zarrinkar, A quantitative analysis of kinase inhibitor selectivity, Nat Biotechnol 26 (2008) 127-132
75. Z.A. Knight, K.M. Shokat, Features of selective kinase inhibitors, Chem Biol 12 (2005) 621-637
76. M.A. Fabian, W.H. Biggs, 3rd, D.K. Treiber, C.E. Atteridge, M.D. Azimioara, M.G. Benedetti, T.A. Carter, P. Ciceri, P.T. Edeen, M. Floyd, J.M. Ford, M. Galvin, J.L. Gerlach, R.M. Grotzfeld, S. Herrgard, D.E. Insko, M.A. Insko, A.G. Lai, J.M. Lelias, S.A. Mehta, Z.V. Milanov, A.M. Velasco, L.M. Wodicka, H.K. Patel, P.P. Zarrinkar, D.J. Lockhart, A small molecule-kinase interaction map for clinical kinase inhibitors, Nat Biotechnol 23 (2005) 329-336
77. M.I. Davis, J.P. Hunt, S. Herrgard, P. Ciceri, L.M. Wodicka, G. Pallares, M. Hocker, D.K. Treiber, P.P. Zarrinkar, Comprehensive analysis of kinase inhibitor selectivity, Nat Biotechnol 29 (2011) 1046-1051
78. T. Anastassiadis, S.W. Deacon, K. Devarajan, H. Ma, J.R. Peterson, Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity, Nat Biotechnol 29 (2011) 1039-1045
79. K. West, CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders, Curr Opin Investig Drugs 10 (2009) 491-504
80. A. Converso, T. Hartingh, R.M. Garbaccio, E. Tasber, K. Rickert, M.E. Fraley, Y. Yan, C. Kreatsoulas, S. Stirdivant, B. Drakas, E.S. Walsh, K. Hamilton, C.A. Buser, X. Mao, M.T. Abrams, S.C. Beck, W. Tao, R. Lobell, L. Sepp-Lorenzino, J. Zugay-Murphy, V. Sardana, S.K. Munshi, S.M. Jezequel- Sur, P.D. Zuck, G.D. Hartman, Development of thioquinazolinones, allosteric Chk1 kinase inhibitors, Bioorg Med Chem Lett 19 (2009) 1240-1244
81. H. Tecle, J. Shao, Y. Li, M. Kothe, S. Kazmirski, J. Penzotti, Y.H. Ding, J. Ohren, D. Moshinsky, R. Coli, N. Jhawar, E. Bora, S. Jacques-O’Hagan, J. Wu, Beyond the MEKpocket: can current MEK kinase inhibitors be utilized to synthesize novel type III NCKIs? Does the MEK-pocket exist in kinases other than MEK?, Bioorg Med Chem Lett 19 (2009) 226-229
82. T. Fox, J.T. Coll, X. Xie, P.J. Ford, U.A. Germann, M.D. Porter, S. Pazhanisamy, M.A. Fleming, V. Galullo, M.S. Su, K.P. Wilson, A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase, Protein Sci 7 (1998) 2249-2255
83. M. Fricker, P. Lograsso, S. Ellis, N. Wilkie, P. Hunt, S.J. Pollack, Substituting c-Jun N-terminal kinase-3 (JNK3) ATP-binding site amino acid residues with their p38 counterparts affects binding of JNK- and p38-selective inhibitors, Arch Biochem Biophys 438 (2005) 195-205
84. M. Kothe, D. Kohls, S. Low, R. Coli, A.C. Cheng, S.L. Jacques, T.L. Johnson, C. Lewis, C. Loh, J. Nonomiya, A.L. Sheils, K.A. Verdries, T.A. Wynn, C. Kuhn, Y.H. Ding, Structure of the catalytic domain of human polo-like kinase 1, Biochemistry 46 (2007) 5960-5971
85. S.F. Bellon, P. Kaplan-Lefko, Y. Yang, Y. Zhang, J. Moriguchi, K. Rex, C.W. Johnson, P.E. Rose, A.M. Long, A.B. O’Connor, Y. Gu, A. Coxon, T.S. Kim, A. Tasker, T.L. Burgess, I. Dussault, c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations, J Biol Chem 283 (2008) 2675-2683
86. I. Dussault, S.F. Bellon, c-Met inhibitors with different binding modes: two is better than one, Cell Cycle 7 (2008) 1157-1160
87. E.R. Wood, A.T. Truesdale, O.B. McDonald, D. Yuan, A. Hassell, S.H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K.J. Alligood, D.W. Rusnak, T.M. Gilmer, L. Shewchuk, A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells, Cancer Res 64 (2004) 6652-6659
88. T. Schindler, W. Bornmann, P. Pellicena, W.T. Miller, B. Clarkson, J. Kuriyan, Structural mechanism for STI-571 inhibition of abelson tyrosine kinase, Science 289 (2000) 1938-1942
89. B.K. Albrecht, J.C. Harmange, D. Bauer, L. Berry, C. Bode, A.A. Boezio, A. Chen, D. Choquette, I. Dussault, C. Fridrich, S. Hirai, D. Hoffman, J.F. Larrow, P. Kaplan-Lefko, J. Lin, J. Lohman, A.M. Long, J. Moriguchi, A. O’Connor, M.H. Potashman, M. Reese, K. Rex, A. Siegmund, K. Shah, R. Shimanovich, S.K. Springer, Y. Teffera, Y. Yang, Y. Zhang, S.F. Bellon, Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase, J Med Chem 51 (2008) 2879-2882
90. N.K. Williams, R.S. Bamert, O. Patel, C. Wang, P.M. Walden, A.F. Wilks, E. Fantino, J. Rossjohn, I.S. Lucet, Dissecting specificity in the Janus kinases: the structures of JAKspecific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains, J Mol Biol 387 (2009) 219-232
91. E.L. Kwak, R. Sordella, D.W. Bell, N. Godin-Heymann, R.A. Okimoto, B.W. Brannigan, P.L. Harris, D.R. Driscoll, P. Fidias, T.J. Lynch, S.K. Rabindran, J.P. McGinnis, A. Wissner, S.V. Sharma, K.J. Isselbacher, J. Settleman, D.A. Haber, Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib, Proc Natl Acad Sci U S A 102 (2005) 7665-7670
92. M. Azam, M.A. Seeliger, N.S. Gray, J. Kuriyan, G.Q. Daley, Activation of tyrosine kinases by mutation of the gatekeeper threonine, Nat Struct Mol Biol 15 (2008) 1109-1118
93. N.J. Dibb, S.M. Dilworth, C.D. Mol, Switching on kinases: oncogenic activation of BRAF and the PDGFR family, Nat Rev Cancer 4 (2004) 718-727
94. A.P. Kornev, N.M. Haste, S.S. Taylor, L.F. Eyck, Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism, Proc Natl Acad Sci U S A 103 (2006) 17783-17788
95. H. Kantarjian, F. Giles, L. Wunderle, K. Bhalla, S. O’Brien, B. Wassmann, C. Tanaka, P. Manley, P. Rae, W. Mietlowski, K. Bochinski, A. Hochhaus, J.D. Griffin, D. Hoelzer, M. Albitar, M. Dugan, J. Cortes, L. Alland, O.G. Ottmann, Nilotinib in imatinib-resistant CML and Philadelphia chromosomepositive ALL, N Engl J Med 354 (2006) 2542-2551
96. H. Kantarjian, N.P. Shah, A. Hochhaus, J. Cortes, S. Shah, M. Ayala, B. Moiraghi, Z. Shen, J. Mayer, R. Pasquini, H. Nakamae, F. Huguet, C. Boque, C. Chuah, E. Bleickardt, M.B. Bradley-Garelik, C. Zhu, T. Szatrowski, D. Shapiro, M. Baccarani, Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia, N Engl J Med 362 (2010) 2260-2270
97. C.H. Yun, K.E. Mengwasser, A.V. Toms, M.S. Woo, H. Greulich, K.K. Wong, M. Meyerson, M.J. Eck, The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP, Proc Natl Acad Sci U S A 105 (2008) 2070-2075
98. T. O’Hare, W.C. Shakespeare, X. Zhu, C.A. Eide, V.M. Rivera, F. Wang, L.T. Adrian, T. Zhou, W.S. Huang, Q. Xu, C.A. Metcalf, 3rd, J.W. Tyner, M.M. Loriaux, A.S. Corbin, S. Wardwell, Y. Ning, J.A. Keats, Y. Wang, R. Sundaramoorthi, M. Thomas, D. Zhou, J. Snodgrass, L. Commodore, T.K. Sawyer, D.C. Dalgarno, M.W. Deininger, B.J. Druker, T. Clackson, AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance, Cancer Cell 16 (2009) 401-412
99. M.A. Lemmon, J. Schlessinger, Cell signaling by receptor tyrosine kinases, Cell 141 (2010) 1117-1134
100. M. Huse, J. Kuriyan, The conformational plasticity of protein kinases, Cell 109 (2002) 275-282
101. X. Zhang, J. Gureasko, K. Shen, P.A. Cole, J. Kuriyan, An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor, Cell 125 (2006) 1137-1149
102. V. Gray-Schopfer, C. Wellbrock, R. Marais, Melanoma biology and new targeted therapy, Nature 445 (2007) 851-857
103. G. Bollag, P. Hirth, J. Tsai, J. Zhang, P.N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E.A. Burton, B. Wong, G. Tsang, B.L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K.Y. Zhang, D.R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D’Andrea, A. Koehler, M. Stumm, P.S. Lin, R.J. Lee, J. Grippo, I. Puzanov, K.B. Kim, A. Ribas, G.A. McArthur, J.A. Sosman, P.B. Chapman, K.T. Flaherty, X. Xu, K.L. Nathanson, K. Nolop, Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma, Nature 467 (2010) 596-599
104. P.B. Chapman, A. Hauschild, C. Robert, J.B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, D. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S.J. O’Day, J.A. Sosman, J.M. Kirkwood, A.M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R.J. Lee, K.T. Flaherty, G.A. McArthur, Improved survival with vemurafenib in melanoma with BRAF V600E mutation, N Engl J Med 364 (2011) 2507- 2516
105. K.T. Flaherty, I. Puzanov, K.B. Kim, A. Ribas, G.A. McArthur, J.A. Sosman, P.J. O’Dwyer, R.J. Lee, J.F. Grippo, K. Nolop, P.B. Chapman, Inhibition of mutated, activated BRAF in metastatic melanoma, N Engl J Med 363 (2010) 809-819
106. N. Wagle, C. Emery, M.F. Berger, M.J. Davis, A. Sawyer, P. Pochanard, S.M. Kehoe, C.M. Johannessen, L.E. Macconaill, W.C. Hahn, M. Meyerson, L.A. Garraway, Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling, J Clin Oncol 29 (2011) 3085-3096
107. C.M. Johannessen, J.S. Boehm, S.Y. Kim, S.R. Thomas, L. Wardwell, L.A. Johnson, C.M. Emery, N. Stransky, A.P. Cogdill, J. Barretina, G. Caponigro, H. Hieronymus, R.R. Murray, K. Salehi-Ashtiani, D.E. Hill, M. Vidal, J.J. Zhao, X. Yang, O. Alkan, S. Kim, J.L. Harris, C.J. Wilson, V.E. Myer, P.M. Finan, D.E. Root, T.M. Roberts, T. Golub, K.T. Flaherty, R. Dummer, B.L. Weber, W.R. Sellers, R. Schlegel, J.A. Wargo, W.C. Hahn, L.A. Garraway, COT drives resistance to RAF inhibition through MAP kinase pathway reactivation, Nature 468 (2011) 968-972
108. R. Nazarian, H. Shi, Q. Wang, X. Kong, R.C. Koya, H. Lee, Z. Chen, M.K. Lee, N. Attar, H. Sazegar, T. Chodon, S.F. Nelson, G. McArthur, J.A. Sosman, A. Ribas, R.S. Lo, Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation, Nature 468 (2010) 973-977
109. P.I. Poulikakos, Y. Persaud, M. Janakiraman, X. Kong, C. Ng, G. Moriceau, H. Shi, M. Atefi, B. Titz, M.T. Gabay, M. Salton, K.B. Dahlman, M. Tadi, J.A. Wargo, K.T. Flaherty, M.C. Kelley, T. Misteli, P.B. Chapman, J.A. Sosman, T.G. Graeber, A. Ribas, R.S. Lo, N. Rosen, D.B. Solit, RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E), Nature 480 (2011) 387-390
110. C.M. Udell, T. Rajakulendran, F. Sicheri, M. Therrien, Mechanistic principles of RAF kinase signaling, Cell Mol Life Sci 68 (2011) 553-565
111. P.I. Poulikakos, C. Zhang, G. Bollag, K.M. Shokat, N. Rosen, RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF, Nature 464 (2010) 427-430
112. G. Hatzivassiliou, K. Song, I. Yen, B.J. Brandhuber, D.J. Anderson, R. Alvarado, M.J. Ludlam, D. Stokoe, S.L. Gloor, G. Vigers, T. Morales, I. Aliagas, B. Liu, S. Sideris, K.P. Hoeflich, B.S. Jaiswal, S. Seshagiri, H. Koeppen, M. Belvin, L.S. Friedman, S. Malek, RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth, Nature 464 (2010) 431-435
