

MS-based methods for detection, quantitation and localisation of pharmaceuticals and metabolites in biological samples
Posted: 18 December 2012 |
Mass spectrometry is a powerful, multi-faceted technique capable of analysing pharmaceuticals and their metabolites in biological matrices. Although it is more commonly applied to proteins, peptides and lipids, an increasing number of studies use mass spectrometry based techniques to detect, quantitate and localise pharmaceuticals and their metabolites. The availability of functionally unique ionisation methods and preparative separation options coupled with the specificity and sensitivity of a mass analyser make mass spectrometry an attractive option in pharmaceutical studies involving biofluids and tissue. This review aims to provide a general description of the primary mass spectrometric and preparative steps used to analyse pharmaceuticals in biological systems…
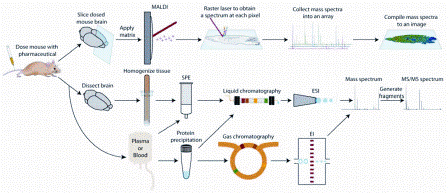
Figure 1: Sample preparation techniques for mass spectrometry based pharmaceutical and metabolite analysis. The three approaches discussed in the paper are outlined: (Top) mass spectrometry imaging from dosed tissue sections, (Middle) liquid chromatography mass spectrometry of tissue extracts or plasma/serum samples, (Bottom) gas chromatography mass spectrometry of plasma/serum samples
Mass spectrometry is a powerful, multi-faceted technique capable of analysing pharmaceuticals and their metabolites in biological matrices. Although it is more commonly applied to proteins, peptides and lipids, an increasing number of studies use mass spectrometry based techniques to detect, quantitate and localise pharmaceuticals and their metabolites. The availability of functionally unique ionisation methods and preparative separation options coupled with the specificity and sensitivity of a mass analyser make mass spectrometry an attractive option in pharmaceutical studies involving biofluids and tissue. This review aims to provide a general description of the primary mass spectrometric and preparative steps used to analyse pharmaceuticals in biological systems.
A leading concern in drug discovery is the potential reactivity of metabolites, which can be biologically transformed from stable pharmaceutical products. Drug metabolites are often considered to be potentially toxic because of their ability to covalently modify proteins and DNA. For this reason, pharmaceutical developers create methods to assess the propensity of new drugs to degrade into reactive metabolites and gauge the effects these metabolites have1. Another area of study, pharmaceutical metabolomics, typically compares the endogenous metabolites of control specimens with drugadministered specimens2. Mass spectrometry has emerged as a useful tool in these two areas because of its throughput, sensitivity and ability to identify multiple molecules in biological media.
Mass spectrometry’s underlying principle is that gaseous ions are detected by a mass analyser according to their mass to charge (m/z) ratio. Therefore, a molecule must become a gaseous ion before it can be detected. Electrospray ionisation (ESI) and matrix-assisted laser desorption / ionisation (MALDI) are the two prevalent ionisation methods, especially for large biomolecules. However, prior to the advent of ESI and MALDI, electron ionisation (EI) has long been used in multiple pharmaceutical studies. An ESI source disperses aqueous analytes into a fine aerosol, utilising solvent evaporation from a charged droplet until becoming unstable and producing multiple and single charged gasphase ions. In MALDI, a weak acid ‘matrix’ extracts and co-crystallises analytes. Upon laser irradiation, matrix and analytes are desorbed into a hot plume. Single charged gas-phase ions form in this plume. Electron impact produces an accelerated stream of electrons concentrated perpendicular to the analyte stream. The close proximity of the electron beam to the neutral molecules induces ionisation and fragmentation.
Analytes are directed to a mass analyser for identification after ionisation. Metabolites are detected in the lower m/z range where interference from other small molecules is possible. Because of this, mass analysers capable of high resolution and accurate mass measurements are preferable. Tandem mass spectrometry (MS/MS) can also assist in identification by fragmenting precursor ions to form product ions unique to the precursor. Common mass analysers in pharmaceutical metabolomics studies include Time-of-Flight/ Time-of-Flight (TOF/TOF), quadrupole-TOF (Q-TOF), triple quadrupole (QqQ), Fourier transform ion cyclotron resonance (FT-ICR) and Orbitrap. Time-of-Flight analysers have a higher acquisition speed (> 20 Hz) but a lower mass accuracy than Orbitrap and FT-ICR instruments (< 2 ppm)2.
Mass spectrometers are generally com – bined with a fractionation method when analysing pharmaceuticals and metabolites in biological samples. Liquid chromatography (LC) and to a lesser extent, gas chromatography (GC), are employed to separate drugs and metabolites before mass spectrometric analysis. Another method, mass spectrometry imaging (MSI), does not utilise a separation step and acquires spatial distribution data of analytes directly from tissue. These three methods are outlined in Figure 1. This review’s purpose is to provide the reader with general descriptions of LC-MS, GC-MS and MSI methods for analysing pharmaceuticals and metabolites in biological samples but is not all-inclusive due to its intended brevity.
Liquid chromatography mass spectrometry (LC-MS)
Ultra-performance liquid chromatography (UPLC) is the preferred method for fractionating pharmaceuticals and metabolites in biological matrices like plasma3, serum4, urine5 and tissue homogenates6. It is relatively easy to couple with ESI and typically uses reversed phase (RP) chromatography to separate dissolved molecules according to their hydrophobic interaction with a solid stationary phase. Common mobile phase solvents for RP-UPLC that are mass spectrometer friendly include combinations of water and methanol / acetonitrile with a volatile acid like formic acid7. Hydrophilic interaction chromatography (HILIC) is another LC method that is increasingly used for pharmaceutical studies8. Hydrophilic interaction chromatography retains very polar, hydrophilic drugs and metabolites that sometimes elute with matrix interferences when RP-UPLC is used, but it has not yet reached the widespread acceptance that RP-UPLC has enjoyed9.
Preparative steps must be undertaken when analysing biological samples before injection onto an LC column. As shown in Figure 1, tissue samples are first homogenised and centrifuged. Solvent extraction and solid phase extraction (SPE) can be used to separate the analytes from remaining matrix interferences in the supernatant10. Protein precipitation11 or SPE12 is typically used before injecting plasma or serum samples while urine is simply diluted13,14.
Figure 1: Sample preparation techniques for mass spectrometry based pharmaceutical and metabolite analysis. The three approaches discussed in the paper are outlined: (Top) mass spectrometry imaging from dosed tissue sections, (Middle) liquid chromatography mass spectrometry of tissue extracts or plasma/serum samples, (Bottom) gas chromatography mass spectrometry of plasma/serum samples
Liquid chromatography mass spectrometry is used extensively in pharmaceutical metabolomics to determine qualitative and quantitative differences in the metabolite profiles of a biological specimen upon administration of a pharmaceutical. This type of work is known as non-targeted metabolomics, and its goal is to identify all detectable compounds and determine which ones change significantly upon an induced stress. Changes in endogenous metabolites are mapped to pathways, revealing mechanistic data about the studied drug2. One example investigated the toxicity of 2,3,7,8-tetrachlorodibenzo-pdioxin (TCDD) in aryl hydrocarbon receptor (AhR) sensitive C57BL/6J and less-sensitive DBA/2J mice15. In this study, mouse strains were orally administered TCDD or vehicle for three consecutive days before sacrifice on the seventh day. Serum samples were analysed via LC-MS/MS in data dependent acquisition (DDA) mode, where precursor ions that had already been selected for MS/MS were excluded for 60 seconds in order to select lower abundance ions. Although many metabolites were unidentified due to the limited size of current databases, 12 differentiating, identified metabolites in C57BL/6J and two in DBA/2J mice were found (P ≤ 0.001 and fold-change ≥ 1.5). As expected, TCDD exhibited greater toxicity through AhR binding in the C57BL/6J mice than the DBA/2J mice. Although the study produced positive results, it highlights that compound identification is a significant challenge in nontargeted metabolomics15.
Quantitative LC-MS/MS analysis, which typically incorporates a reference or isotopic internal standard for each analyte, is becoming quite popular in drug and metabolite studies. One such study developed an LC-MS/MS assay for the simultaneous determination and quantitation of naloxone (NLX) and its two metabolites, 6β-naloxol (NLL) and naloxone-3β- D-glucuronide (NLG), in mouse plasma16. While NLX reverses opioid-induced depression following surgery, NLL and NLG may have toxic properties. Therefore, deuterated internal standards were added to mouse plasma samples containing NLX, NLL and NLG. Multiple reaction monitoring (MRM), which enables quantitation by monitoring precursor to product ion transitions, was performed via a RP-HPLC QqQ system. This method was found to be rapid, specific and reliable, and it is an example of mass spectrometry’s promise in pharmacokinetic studies16.
Gas chromatography mass spectrometry (GC-MS)
The contribution of LC-MS to pharmaceutical and metabolomic analyses cannot be overstated, but GC-MS is an older, wellestablished technique that is also frequently used in this area. A gaseous mobile phase functions as a carrier gas for analytes that partition between the mobile phase and the liquid stationary phase according to boiling point. Electron impact produces ion fragments useful for identification through commercial or in-house library matching2. Unfortunately, an EI spectrum cannot be used to identify metabolites without library matching, and library searches do not identify all signals in a GC-MS chromatogram. Because of this, atmospheric pressure chemical ionisation (APCI) is sometimes used to provide accurate masses of unknown molecular ions17. Another drawback is that an extra step is added to sample preparation. Analytes must be derivatised via acylation18 or silylation19 to increase volatility. Despite these limitations, pharmaceutical and metabolomic studies increasingly utilise GC-MS because of the high resolution of capillary columns and existence of EI libraries.
One impressive GC-MS study investigated quantitative alterations of endogenous metabolites during the development and treatment of type 2 diabetes mellitus (T2DM)20. This experiment’s goal was to determine metabolic changes stemming from T2DM and its drug treatments in a sample pool of 74 patients. Each individual had been diagnosed with T2DM and treated for 48 weeks with repaglinide, metformin or rosiglitazone. The GC-MS analysis protocol used was able to detect 212 individual metabolites consistently in 90 per cent of the human serum samples, but only 67 were positively identified through library references before multivariate data analysis. Significant variations (P < 0.05) of metabolites in diabetic subjects compared to healthy individuals included increases of valine, maltose, glutamate, urate, butanoate, and long chain fatty acid (C16:0, C18:1, C18:0, octadecanoate, and arachidonate) and decreases of glucuronolactone, lysine and lactate20. Each treatment down-regulated high glutamate levels in T2DM patients, but rosiglitazone reversed abnormal metabolite levels the most20. This result suggested that of the three drugs studied, rosiglitazone alters the metabolism of T2DM patients most efficiently.
Another interesting study developed a GCMS method to quantitate buprenorphine (BPN), a semi-synthetic opioid agonist-antagonist and its metabolite nor-buprenorphine (nor-BPN) in blood21. After the addition of a deuterated internal standard, SPE and derivatisation, this GC-MS method was able to assay BPN and nor-BPN with a linear dynamic range of 0.05 – 30.0 μg/L. Since BPN and nor-BPN levels are relatively low in most clinical cases, the method was determined to be applicable. Furthermore, the method could be used in forensic investigations since it is developed for use with blood rather than plasma21.
Mass spectrometry imaging (MSI)
Whole-body autoradiography (WBA) is a wellestablished technique capable of determining the spatial distribution of radiolabeled pharmaceuticals and metabolites inside of or on a biological specimen’s surface. Unfortunately, this method is unable to distinguish parent compound from metabolite distributions. While coupling WBA with LC-MS of tissue extracts is possible, MSI has become a popular alternative due to its ability to obtain spatial distribution data with the molecular specificity of mass spectrometry22. Mass spectrometry imaging consists of rastering a laser or other ionisation source across a thin tissue section by moving the tissue in a grid-like pattern to generate thousands of position-dependent mass spectra. Mass spectra are then assembled so that the spatial distributions of molecules belonging to specific m/z ratios can be displayed as colourcoded ion intensity maps. This method’s ability to perform label-free detection of multiple molecules simultaneously in complex samples elevates it above pre-existing molecular imaging platforms.
The primary ionisation technique in pharmaceutical MSI studies is MALDI. Successful desorption and ionisation of compounds depends on appropriate sample preparation and matrix application. After tissue is harvested from dosed animals, it is immediately snap frozen and stored at -80°C. A cryostat slices tissue into thin sections (3-50 μm), and these sections are then mounted onto a conductive stainless steel plate or indium-tin oxide (ITO)- coated glass slide before freeze-drying and storing at -80°C until matrix application. Common matrices in drug and metabolite studies are 2,5-dihydroxybenzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (CHCA), but matrix selection often involves testing several matrices to find one that does not introduce interfering matrix ions. Matrix deposition should result in homogenous, tiny crystals. Application methods include pneumatic air sprayers23, commercial matrix applicators24, sublimation25, dry-coating26 and homemade nanospotters27.
Many pharmaceutical therapies are tailored to target biological processes in specific organ and tissue compartments, necessitating the need for MSI determinations of locally administered drugs within tissue sites of targeted action. One such proof-of-principle study measured the occurrence of an inhaled bronchodilator, ipratropium, in human bronchial biopsies obtained by fiber optic bronchoscopy28. Biopsies were harvested shortly after dosing in five subjects who were being evaluated for airway obstruction or potential tumour development. Biopsy sections 10 μm thick were coated with CHCA and imaged with a MALDI LTQ Orbitrap XL mass spectrometer. Drug ion signals were also registered to biopsy microstructures by overlaying the MSI data with histological images obtained from hematoxylin and eosin (H&E) staining. The study showed that ipratropium is rapidly absorbed into the airway wall with estimated concentrations between three and 80 nM throughout the wall28. More importantly, it also demonstrated the use of a mass spectrometer with high mass spectral resolution and accuracy to detect and image relevant drugs at a clinical dose within human biopsy material. High resolution instruments like the FT-ICR and Orbitrap from this study can unambiguously detect drugs and metabolites even in the presence of matrix interference because of their ability to resolve analytes from interfering ions with the same nominal mass29. Unfortunately, these mass analysers generally take longer to construct an image because they acquire spectra much slower than lower resolution instruments.
A few challenges exist when using a matrix to assist with desorption and ionisation. Because analytes are extracted into matrix crystals, spatial resolution is dependent on both the laser beam width and crystal size. Many pharmaceutical MALDI-MSI studies report lateral resolutions of about 100 μm, which is too poor to elucidate some tissue microstructures. Secondary ion mass spectrometry (SIMS) is capable of submicron resolutions, but its hard ionisation mechanism and lack of commercially available MS/MS capabilities have limited its use30. Several matrix-free approaches in the nascent development stage include nano – structure initiator mass spectrometry (NIMS)31, desorption electrospray ionisation (DESI)32, and laser ablation electrospray ionisation (LAESI)33, but none of these methods have demonstrated vastly improved spatial resolutions. One MALDI variant, scanning microprobe MALDI (SMALDI), has successfully generated ion maps in biological tissue with ~5 μm resolution34. This method uses laser prefocusing and final focusing steps with suprasil quartz lenses to achieve greater lateral resolutions than MALDI is typically capable of. The SMALDI method recently analysed the spatial distributions of anti-cancer drugs imatinib and ifosfamide in mouse organs at 10 μm35. Although this modification has not replaced traditional MALDI sources, developments like this example guarantee MSI’s continued use in pharmaceutical and metabolite studies.
Conclusion
A variety of mass spectrometric methods are used in pharmaceutical and metabolite analyses. LC- and GC-MS are both used to survey the metabolome after drug administration as well as monitor the tendency of a drug to be transformed into reactive metabolites. Fast instruments with high resolution and larger, more diverse chemical libraries for compound identification are improving the validity and quantity of data per experiment. While MSI is a relatively new technology, it is featured in many studies focusing on drug and metabolite localisation due to its ability to perform labelfree analyses of multiple molecules in tissue. High repetition rate lasers, continuous laser rastering, and self-cleaning sources have improved MSI’s throughput and robustness. Tandem mass analysers and high resolution instruments have also improved confidence in MSI measurements. Continuous development in each mass spectrometry technique discussed ensures that current challenges and limitations will soon be addressed, cementing mass spectrometry’s role in the future of pharmaceutical studies.
Acknowledgement
Preparation of this review was supported in part by National Science Foundation (CHE- 0957784). L.L. acknowledges an H. I. Romnes Faculty Fellowship.
References
- Ma, S.; Zhu, M. Chem Biol Interact 2009, 179, 25-37
- Drexler, D. M.; Reily, M. D.; Shipkova, P. A. Anal Bioanal Chem 2011, 399, 2645-2653
- Rodamer, M.; Elsinghorst, P. W.; Kinzig, M.; Gutschow, M.; Sorgel, F. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879, 695-706
- Teunissen, S. F.; Jager, N. G.; Rosing, H.; Schinkel, A. H.; Schellens, J. H.; Beijnen, J. H. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879, 1677-1685
- Herebian, D.; Lamshoft, M.; Mayatepek, E.; Spiekerkoetter, U. Rapid Commun Mass Spectrom 2010, 24, 791-800
- Miyazono, Y.; Harada, K.; Sugiyama, K.; Ueno, M.; Torii, M.; Kato, I.; Matsuura, H.; Hirata, K. J Appl Toxicol 2011, 31, 655-662
- Hsieh, Y.; Korfmacher, W. Curr Pharm Des 2009, 15, 2251-2261
- Song, Q.; Naidong, W. J Chromatogr B Analyt Technol Biomed Life Sci 2006, 830, 135-142
- Dejaegher, B.; Vander Heyden, Y. J Sep Sci 2010, 33, 698-715 10. Zhou, Q.; Gallo, J. M. J Pharm Biomed Anal 2010, 51, 958-964
- Huang, J.; Gautam, N.; Bathena, S. P.; Roy, U.; McMillan, J.; Gendelman, H. E.; Alnouti, Y. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879, 2332-2338
- Bhatt, M.; Shah, S. Biomed Chromatogr 2010, 24, 1247-1254
- Lutz, U.; Bittner, N.; Lutz, R. W.; Lutz, W. K. J Chromatogr B Analyt Technol Biomed Life Sci 2008, 871, 349-356
- Sun, J.; Von Tungeln, L. S.; Hines, W.; Beger, R. D. J Chromatogr B Analyt Technol Biomed Life Sci 2009, 877, 2557-2565
- Lin, S. H.; Yang, Z.; Shen, Y.; Cai, Z. W. Int J Mass Spectrom 2011, 301, 29-36
- Jiang, H.; Wang, Y.; Shet, M. S.; Zhang, Y.; Zenke, D.; Fast, D. M. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879, 2663-2668
- Wachsmuth, C. J.; Almstetter, M. F.; Waldhier, M. C.; Gruber, M. A.; Nurnberger, N.; Oefner, P. J.; Dettmer, K. Anal Chem 2011, 83, 7514-7522
- Khraiwesh, A.; Papoutsis, I.; Nikolaou, P.; Pistos, C.; Spiliopoulou, C.; Athanaselis, S. J Chromatogr B Analyt Technol Biomed Life Sci 2011, 879, 2576-2582
- Mullot, J. U.; Karolak, S.; Fontova, A.; Huart, B.; Levi, Y. Anal Bioanal Chem 2009, 394, 2203-2212
- Bao, Y.; Zhao, T.; Wang, X.; Qiu, Y.; Su, M.; Jia, W. J Proteome Res 2009, 8, 1623-1630
- Papoutsis, II; Nikolaou, P. D.; Athanaselis, S. A.; Pistos, C. M.; Spiliopoulou, C. A.; Maravelias, C. P. J Pharm Biomed Anal 2011, 54, 588-591
- Greer, T.; Sturm, R.; Li, L. J Proteomics 2011, 74, 2617-2631
- Acquadro, E.; Cabella, C.; Ghiani, S.; Miragoli, L.; Bucci, E. M.; Corpillo, D. Anal Chem 2009, 81, 2779-2784
- Nilsson, A.; Fehniger, T. E.; Gustavsson, L.; Andersson, M.; Kenne, K.; Marko-Varga, G.; Andren, P. E. PLoS One 2010, 5, e11411
- Dekker, L. J.; van Kampen, J. J.; Reedijk, M. L.; Burgers, P. C.; Gruters, R. A.; Osterhaus, A. D.; Luider, T. M. Rapid Commun Mass Spectrom 2009, 23, 1183-1188
- Goodwin, R. J.; Macintyre, L.; Watson, D. G.; Scullion, S. P.; Pitt, A. R. Rapid Commun Mass Spectrom 2010, 24, 1682-1686
- Vegvari, A.; Fehniger, T. E.; Gustavsson, L.; Nilsson, A.; Andren, P. E.; Kenne, K.; Nilsson, J.; Laurell, T.; Marko- Varga, G. J Proteomics 2010, 73, 1270-1278
- Fehniger, T. E.; Vegvari, A.; Rezeli, M.; Prikk, K.; Ross, P.; Dahlback, M.; Edula, G.; Sepper, R.; Marko-Varga, G. Anal Chem 2011, 83, 8329-8336
- Castellino, S.; Groseclose, M. R.; Wagner, D. Bioanalysis 2011, 3, 2427-2441
- Chehade, F.; de Labriolle-Vaylet, C.; Moins, N.; Moreau, M. F.; Papon, J.; Labarre, P.; Galle, P.; Veyre, A.; Hindie, E. J Nucl Med 2005, 46, 1701-1706
- Northen, T. R.; Yanes, O.; Northen, M. T.; Marrinucci, D.; Uritboonthai, W.; Apon, J.; Golledge, S. L.; Nordstrom, A.; Siuzdak, G. Nature 2007, 449, 1033-1036
- Takats, Z.; Wiseman, J. M.; Gologan, B.; Cooks, R. G. Science 2004, 306, 471-473.
- Nemes, P.; Vertes, A. Anal Chem 2007, 79, 8098-8106
- Rompp, A.; Guenther, S.; Schober, Y.; Schulz, O.; Takats, Z.; Kummer, W.; Spengler, B. Angew Chem Int Ed Engl 2010, 49, 3834-3838
- Rompp, A.; Guenther, S.; Takats, Z.; Spengler, B. Anal Bioanal Chem 2011, 401, 65-73
About the authors
Tyler Greer is a PhD graduate student in the Analytical Chemistry program at University of Wisconsin-Madison. He currently investigates mass spectrometry imaging development and protein and peptide quantitation strategies in Lingjun Li’s group. He received his BSc in Chemistry from University of Michigan in 2009.
Dr. Lingjun Li obtained her PhD degree in Analytical Chemistry / Biomolecular Chemistry in 2000 at the University of Illinois at Urbana-Champaign (UIUC). She then did a three-way postdoctoral research at the Pacific Northwest National Laboratory, Brandeis University, and UIUC before starting her independent faculty position at the University of Wisconsin-Madison (UW-Madison) in December 2002. Dr. Li is currently Professor of Pharmaceutical Sciences and Chemistry at UW-Madison. Her research interests are in analytical neurochemistry, neuropeptidomics and biological mass spectrometry (MS). Her research program focuses on developing novel MS tools such as imaging MS, chemical labelling and quantitation strategies coupled with microseparation techniques to study challenging neuroscience problems with an emphasis on neuropeptides and biomarker discovery in neurodegenerative diseases.

