Coupling large molecules with targeting and delivery mechanisms to augment intracellular delivery
Posted: 21 February 2022 | Guy Hermans (Sapreme Technologies), Miriam Bujny (Sapreme Technologies) | No comments yet
Most intracellular drug targets remain intractable due to cellular membranes effectively denying large foreign molecules entry into the cytoplasm. Over recent decades, several technologies that enable biologics to enter cells have emerged, but successes remain limited to date. Here, Sapreme Technologies’ Miriam Bujny, Chief Development Officer, and Guy Hermans, Chief Executive Officer, discuss novel bioconjugation methods to incorporate specific cell-targeting and membrane-penetrating mechanisms into therapeutics to effectively engage previously elusive intracellular targets.

A range of approaches in modern biotechnology necessitate the safe and efficient intracellular delivery of therapeutic cargo such as proteins, oligonucleotides and small molecules to ensure therapeutic effect. However, tissues and their constituent cells have evolved mechanisms and physical barriers to prevent foreign molecules from entering the cellular cytoplasm, thus blocking the delivery of therapeutics. In the event that a large molecular drug succeeds in reaching the target cells and becomes internalised, for many protein and gene delivery applications such uptake is largely non-productive as a significant portion remains trapped in the cells’ internal membrane systems and ends up in degradative pathways. It is estimated that only a minor fraction of protein and less than one percent of a given oligonucleotide1 manages to escape and be released into the cytosol. Accordingly, only a very minor fraction of the administered drug is ultimately available to exert any pharmacologically relevant effect, often insufficient to afford a useful therapeutic window. To unlock the full potential of promising novel therapeutic modalities for intracellular targets, the development of effective, clinically relevant cell-targeting and cytoplasmic delivery mechanisms is a key challenge.
Large molecule internalisation and endosomal escape inspired by nature
In principle, two major pathways exist for large molecules to reach the inside of a cell: by direct penetration of the plasma membrane or by hijacking cellular internalisation pathways. The most common internalisation pathway is uptake by receptor-mediated endocytosis. Direct traversal of the plasma membrane is virtually impossible for large molecules given their size, charge, and the biophysical properties of the cell membrane. Mechanisms to escape the confines of the endosomal pathways following internalisation, however, have evolved in several organisms – for example, in viruses, bacteria and in the form of plant secondary metabolites – equipping these organisms with means to gain complete entry into the cytosol of a host or predator cell, or insertion of specific payloads into the same. In the context of drug development, targeting endosomal uptake and engineering endosomal escape approaches based on these mechanisms has become an attractive concept for intracellular delivery of large drug molecules.
Conjugation for cell-specific targeting or cell membrane penetration
A relatively recent development is to equip large drug molecules like proteins and oligonucleotides with cell-specific targeting and internalising mechanisms through conjugation, routing them into the cells’ endosomal pathway. This enables the cargo to become internalised in target cells and then traverse the endosomal membranes to reach the cytoplasm or nucleus. If internalised therapeutic modalities do not escape in time, they will be enzymatically degraded in the lysosomes. Thus, incorporation of effective cell membrane‑penetrating mechanisms has the potential to drastically improve drug delivery to the cytoplasm, especially if those modalities can mediate escape from the early or late endosomal compartments. Other approaches, including lipid conjugations, formulation in lipid nanoparticles or DNA caging, have recently been reviewed2 and are excluded from this article.
Cell-specific targeting
Conjugating targeting moieties allows for directing the cargo to select cells and routing them to the endosomal compartment. This approach is now common in antibody-drug conjugates (ADC), developed mainly as oncology therapeutics. The prototypical ADC consists of a monoclonal antibody attached to a small-molecule cytotoxic payload, where the antibody serves to recognise and bind a protein overexpressed or exclusively expressed on the tumour cell surface. In essence, the compound then hitches a ride into the target cells using the receptor’s intrinsic internalisation route and is delivered to an intracellular compartment from which it is ultimately released into the cytoplasm. More recently, these concepts have also been translated in non-oncology applications, with several ligand-conjugated oligonucleotides reaching the market or in late‑stage clinical trials (see Table 1). 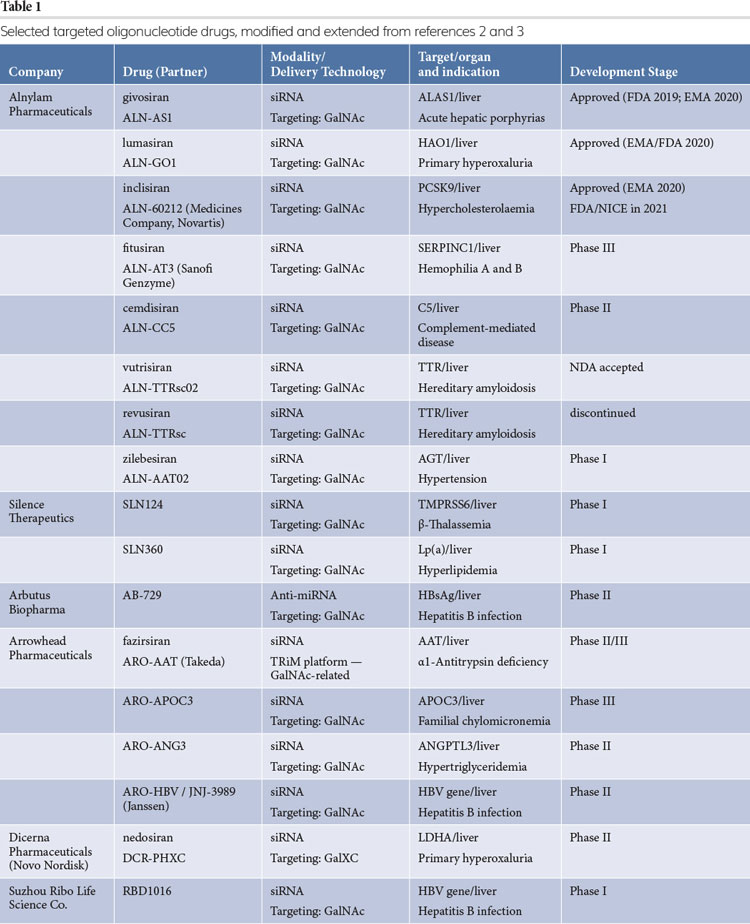 To date, the vast majority of ligand-oligonucleotide conjugates on the market or in advanced clinical development are designed for liver targeting, employing N-acetyl-galactosamine (GalNAc). GalNAc is a ligand of the highly expressed hepatocyte-associated asialoglycoprotein receptor (ASGPR) that internalises circulating glycoproteins via receptor-mediated endocytosis and continuously recycles back to the plasma membrane (Figure 1).4 While GalNAc-conjugation has been transformative for improving tissue-specific delivery, internalisation and efficacy of small interfering RNA (siRNA) therapeutics, extrahepatic-targeted delivery has remained a major challenge.
To date, the vast majority of ligand-oligonucleotide conjugates on the market or in advanced clinical development are designed for liver targeting, employing N-acetyl-galactosamine (GalNAc). GalNAc is a ligand of the highly expressed hepatocyte-associated asialoglycoprotein receptor (ASGPR) that internalises circulating glycoproteins via receptor-mediated endocytosis and continuously recycles back to the plasma membrane (Figure 1).4 While GalNAc-conjugation has been transformative for improving tissue-specific delivery, internalisation and efficacy of small interfering RNA (siRNA) therapeutics, extrahepatic-targeted delivery has remained a major challenge.
Cell membrane penetration
Direct conjugation of moieties that mediate lipid membrane penetration, such as cell penetrating peptides (CPPs) or small molecular weight glycosylated triterpenoid-based endosomal escape enhancers, can provide efficient means to traverse cellular membranes.
CPPs are short peptide sequences, typically α-helical with a high overall cationic charge, that can be conjugated to a drug molecule or may be genetically encoded as a fusion in the protein- and peptide-drug context. The assumed mechanism of action is that direct electrostatic interaction with negatively charged phospholipids of the cell membranes enables the translocation of even large molecules via an energy-independent process across membranes.5 The prototypical CPP is the HIV-1-encoded TAT, which was the first CPP discovered. Since then, CPPs have been explored in intracellular drug delivery to target intracellular protein-protein interactions, especially in oncological applications up to evaluation in Phase II clinical trials. A novel wave of optimised CPPs found its way in indications in oncology and beyond.
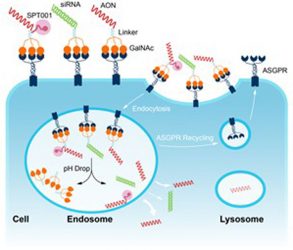
Figure 1: GalNAc-mediated siRNA and antisense oligonucleotide (AON) delivery to ASGPR-expressing hepatocytes; modified from reference 4.
One inherent drawback of CPPs that directly penetrate the plasma membrane is their lack of cell/tissue specificity, which can lead to possible side effects, including averse immunogenic reactions. Ideally, a delivery mechanism should combine both cell- or tissue-specific delivery with efficient cytoplasmic or delivery through endosomal escape in the selected cell type only. For such a combined approach, a promising class of endosomal escape enhancers consists of engineered glycosylated triterpenoids. Glycosylated triterpenoids are secondary plant metabolites and several have been successfully used in research, particularly in the context of immuno-oncology, to deliver antibody-targeted anti-tumour toxins lacking an intrinsic membrane translation module. Several studies in mice demonstrated that the use of such glycosylated triterpenoids results in improved efficacy of anti-tumour therapy. For instance, co‑administration of the metabolite and a targeted toxin resulted in 94 percent tumour volume reduction and complete regression in seven out of 10 animals,6 whereas the targeted toxin alone resulted in only a 42 percent reduction. Even when the monotherapy dose was increased 50-fold, it still only resulted in 71 percent tumour reduction.6 Intriguingly, such glycosylated triterpenoids act as pH-sensitive release agents, which affords them a high degree of specificity to mediate release only in an environment with suitable conditions, such as late endosomes or endolysosomes. Until recently, their application was limited to co-administration settings, which hampered their applicability in the drug development context. Structured medicinal chemistry-type campaigns and functional high‑throughput screening efforts have now yielded several glycosylated triterpenoids that show a high degree of pH dependency and thus compartmental specificity, providing an excellent balance of safety in circulation with good endosomal escape enhancement properties. These optimised molecules can readily be conjugated to targeted large molecules using various proven linker chemistries such as those already optimised in the context of ADCs or antibody-oligo conjugates. As a frontrunner in these methods, Sapreme Technologies has published in vivo proof-of-concept data showing marked potency improvement for glycosylated triterpenoids (SPT001) conjugated GalNAc-oligonucleotides over both non-targeted oligonucleotide or GalNAc-oligonucleotide without escape enhancer in the same model.7 Further developments expanding the technology to extrahepatic tissue-delivering ligands and other payloads are well underway.
Outlook
The direct conjugation of targeting moieties to large molecule drugs has delivered several successful therapies for efficient hepatic delivery of oligonucleotides. Extrahepatic targeting remains challenging, but promising results for tumour cell delivery and indications beyond are becoming apparent. Beyond this, novel approaches combining cell-specific targeting with endosomal escape enhancement have recently emerged and are rapidly advancing through pre-clinical evaluation. Such combined approaches have the potential to markedly increase on-target delivery in hepatic and extra-hepatic tissues and make previously intractable intracellular targets druggable. While still in its infancy, endosomal escape enhancers such as Sapreme Technologies’ SPT001 have the potential to make formerly undeliverable modalities viable therapeutic options in the future.
Miriam Bujny is Chief Development Officer at Sapreme Technologies. She has over 10 years of experience in drug discovery, translational science and early-stage clinical development. Miriam holds a PhD in biochemistry from the University of Bristol with a specialisation in endosomal transport and how bacteria and bacterial toxins hijack endosomal pathways. She completed postdoctoral training in the lab of Dr Xiaowei Zhuang at Harvard University, specialising in developing imaging methods for application to biomedical questions, such as viral infections. Guy Hermans joined Sapreme Technologies as Chief Executive Officer in January 2020. Prior to this, he held the position of Chief Scientific Officer at Isogenica Ltd., where he was responsible for refocusing the company as an antibody discovery technology provider and improving profitability. Before Isogenica, he held multiple senior research positions at Ablynx N.V. between 2003 and 2015. Guy holds a PhD in medicine from UHasselt and completed his postdoc at Stanford University School of Medicine.
References
- Dowdy S. Overcoming cellular barriers for RNA therapeutics. Nature Biotechnology. 2017 Mar; 25(3): 222–229.
- Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nature Reviews Drug Discovery. 2020 Aug;19:673 – 694.
- Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020 Jun 19;5(1):101.
- Cui H, Zhu X, Li S, et al. Liver-Targeted Delivery of Oligonucleotides with N-Acetylgalactosamine Conjugation. ACS Omega. 2021 Jun;6(25):16259-16265. Figure 1. GalNAc-mediated siRNA and ASO delivery to liver hepatocytes.
- Habault J, Poyet JL. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules. 2019 Mar;24(5):927.
- Bachran C, Dürkop H, Sutherland M, et al. Inhibition of tumor growth by targeted toxins in mice is dramatically improved by saponinum album in a synergistic way. Journal of Immunotherapy. 2009 Sep; 32(7), 713-725.
- Sapreme Unveils its Proprietary Endosomal Escape Platform in Presentations at 16th Annual Meeting of Oligonucleotide Therapeutics Society [Internet]. BusinessWire.BusinessWire; 2020 [cited 2022Jan14]. Available from: https://www.businesswire.com/news/home/20200928005040/en/Sapreme-Unveils-its-Proprietary-Endosomal-Escape-Platform-in-Presentations-at-16th-Annual-Meeting-of-Oligonucleotide-Therapeutics-Society