

Getting to grips with drug resistance in the human protein kinase superfamily
Posted: 21 February 2013 |
Protein kinases represent a vast, partially untapped resource of drug targets for therapeutic intervention in human disease. The remarkable success of the tyrosine kinase inhibitor Imatinib, which is now the first-line therapy in Philadelphia-positive tyrosine kinase inhibitor Imatinibhas galvanised biomedical researchers in an attempt to repeat the landmark success of this ‘bench-to-bedside’ approach to therapy[1]. Imatinib inhibits the BCR-ABL fusion kinases responsible for driving these cancers, and its clinical efficacy provides compelling molecular evidence that this drug elicits life-extending clinical responses through an ‘on-target’ mechanism. Interestingly, Imatinib has several additional (non-ABL) protein kinase targets including oncogenic KIT, which also allows it to be employed for the treatment of high-risk Gastro Intestinal Stromal Tumours…

Re-purposing mechanistically-validated kinase inhibitors with a large medicinal chemistry footprint is an attractive approach for treating diseases whose symptoms are driven by distinct drug-sensitive protein kinases, and this strategy ensures improved returns on scientific and financial investments. However, the remarkable clinical effect of Imatinib is significantly diminished by the appearance of drug-resistant patient sub-populations, which contain point mutations in the ABL kinase domain. These findings, which can be recapitulated experi – mentally, unequivocally validate ABL as a bona fide target of Imatinib, but until recently represented a death-sentence for patients when they appeared[2]. By exploiting the tricks of chemical biology and employing model organismal cell biology, we are now in a robust position to dissect acquired resistance mechanistically and redouble our efforts to predict and override drug resistance in vivo. This response should be driven by a broad understanding of the mechanism(s) by which small molecule kinase inhibitors interact with their targets and by cataloguing the sometimes unpredictable responses of cells to kinase inhibitors. In this article, I discuss findings implicating the kinome-wide mutation of a select group of amino acids, which confer general and/or specific drug-resistance to kinase inhibitors, and highlight how such resistance can be exploited and tackled in the future.
Table 1: Three generations of BCR-ABL inhibitor mentioned in this review
Name and Structure of inhibitor | Generation and year of approval | Kinases targeted |
Imatinib (Gleevec/Glivec)
| 1st Generation 2001 | BCR-ABL, not mutated variants, PDGFR, KIT |
Dasatinib
| 2nd Generation 2006 | Imatinib-refractory BCR-ABL (not T315I), SRC, KIT |
Nilotinib
| 2nd Generation 2007 | Imatinib-refractory BCR-ABL (not T315I), SRC, KIT |
Ponatinib
| 3rd Generation 2012 | BCR-ABL T315I, FLT3, KIT, RET, others |
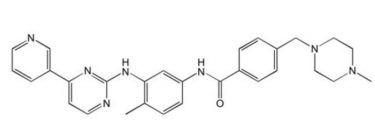
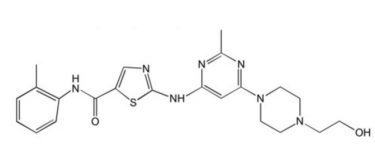
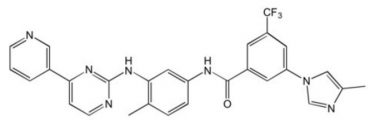
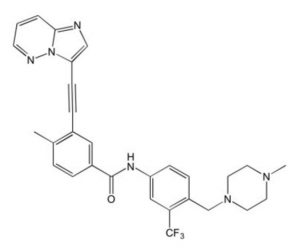
Imatinib is the founding father of the kinase inhibitor field
Protein kinases are a superfamily of enzymes whose central feature is their common ability to bind ATP and (in most cases) catalyse the transfer of the γ-phosphate to amino acid acceptors on protein substrates. These reversible phosphorylation events play a pivotal role in regulating cell signalling under normal and pathological conditions. Indeed, the central ability of protein kinases to control the cell cycle, proliferation and apoptosis marked them out at an early stage as potential molecular targets (and anti-targets) in the cancer field. The protein kinase inhibitor market is projected to become a USD 100 billion global powerhouse in the next decade, as the approval of kinase inhibitors for new cancer indications accelerates and expands to encompass inflammatory and infectious diseases[3]. Remarkably, it is only a decade or so ago since Imatinib (Table 1) became the first kinase inhibitor to reach the market. Since then, a number of new, largely predictable, challenges have arisen and taken centre stage in the field. From a drug-resistance perspective, we are now in a strong position to scrutinise trial and patient data from the first group of kinase inhibitors to enter the clinic, and integrate this information with the vast amount of pre-clinical data compiled from biochemical and cellular screens of kinase inhibitors[4], much of which has yet to enter the public domain. Scrutiny of clinical trial data reveals that in many cancer indications, single agent regimes employing highly targeted kinase inhibitors delivered as single agents often have unacceptably high failure rates. These include an inability to eradicate solid tumours in which the rate of proliferative has subsided (leaving behind a genomically-unstable mixture of cell types), dose-limiting toxicity / side-effects, and the seemingly inevitable selection of cancer cell populations harbouring drug-resistant genotypes. The success of Imatinib as a mono – therapy suggests that drugs targeting the ABL kinase in leukaemia represent an exception to this rule, although the acquired drug-resistance documented in a significant percentage of inhibitor-exposed CML patients can be thought of as an important (but nonetheless unwanted) test bed in which next generation drugs can be assessed. The outcomes observed with Imatinib closely parallel paradigms from bacterial and retroviral systems, in which drug-resistance can appear after exposure to antibiotics or HIV proteinase inhibitors. Consequently, similar lessons being learnt in the cancer clinic should now be integrated with research data and drug development and delivery strategies repositioned to counter the ability of tumours to rapidly outmanoeuvre kinase inhibitors through multifactorial drug-resistance mechanisms. Today, biologists, chemists and clinicians have many of the tools to predict and react to these phenomena in a kinome-wide manner, helping to generate new approaches and procedures to thwart the menace of acquired drug resistance.
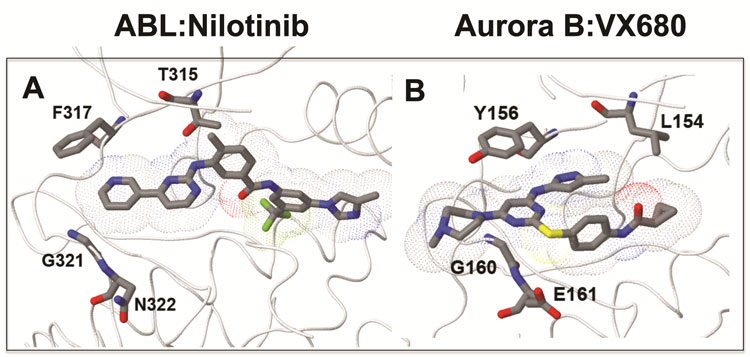
Figure 1 (A) Binding mode and molecular surface of the second generation ABL inhibitor Nilotinib bound to inactive human ABL kinase (PDB ID: 3CS9). (B) Binding mode and molecular surface of the pan-Aurora kinase inhibitor VX680 bound to human Aurora B (PDB ID: 4AF3. In (A), the methylbenzyl moiety engages the pocket adjacent to the gatekeeper (T315) side chain, explaining why Imatinib-resistant BCR-ABL mutations, such as T315I found in CML patients are also resistant to this drug. In (B), VX680 does not engage with the L154 gatekeeper residue, but does bind in the vicinity of the other resistance-tetrad amino acids, explaining how drug-resistance can be produced by their mutation. The four amino acids in the ATP-binding site that make up the ‘resistance-tetrad’ in each kinase are labelled
From Staurosporine to three generations of BCR-ABL kinase inhibitor
The protein kinase superfamily has been targeted for decades by medicinal chemists, although initial efforts had the disadvantage of employing very limited numbers of kinases with which to conduct specificity screening. This often created the false impression that ATP-competitive kinase inhibitors were highly specific cellular tools and that data obtained with drug discovery leads could be interpreted accurately. This situation changed somewhat with the publication of early inhibitor screens using larger panels of protein kinases[5] and the subsequent annotation of the human kinome[6]. A prescient example of this problem is provided by the natural product Staurosporine, a totally non-specific inhibitor of most (but not all) protein kinases that was widely employed as a ‘specific’ PKC inhibitor for many years, before its high levels of promiscuity were appreciated. Indeed, Staurosporine is now used primarily as a tool for inducing cellular apoptosis, rather than as a clinical drug template. That said, chemical efforts at improving the selectivity of Staurosporine have produced useful agents like UCN-01 (7- hydroxystaurosporine) a multikinase inhibitor that has synergic potential as a clinical partner alongside previously licensed agents.
One important lesson learnt with kinase inhibitors might be emphasised: pharmacological agents that inhibit protein kinases need not be (and rarely are) monospecific for one kinase in order to be clinically successful. Indeed, Imatinib has multiple (known) targets, and in some circumstances, simultaneous targeting of multiple protein kinases through ‘polypharmacology’ is likely to be beneficial, either because it has simultaneous anti-proliferative and proapoptotic effects, or because opportunities for the evolution of drug resistance in vivo are minimised[7]. Due to the high degree of similarity exhibited by ‘active’ kinase conformations, which are optimised to trap and hydrolyse the ubiquitous ligand ATP, small molecules that recognise this confomer (the ‘type I’ class, exemplified by Staurosporine) often have multiple targets interspersed throughout the kinome. However, ‘type II’ inhibitors, exemplified by Imatinib, can interact preferentially with the much less-highly conserved ‘inactive’ kinase conformation. Predictably, such inhibitors often exhibit improved specificity profiles, although a significant susceptibility to drug-resistance inducing mutations in their targets is still apparent. In some ways, a clinical lack of requirement for pharmacological ‘monogamy’ is rather fortuitous, because many protein kinase inhibitors were identified and optimised in catalytically active kinase screens[8], although improved cellular assays are helping to reverse this tendency[9]. On the flip-side, the chemical biology community often require highly specific protein kinase inhibitors (validated with drug resistant kinase mutants) to permit accurate interpretation of data when they are employed in cellular or whole animal models[10,11].
The development of clinical inhibitors, and the ground-breaking success of Imatinib, provides a remarkable example of how targeted therapies can become potential curatives once patient stratification and a mixed portfolio of drugs are successfully developed. The key to the success of Imatinib lies in both the cancers where it is employed (predominantly blood-borne) and its cellular targets, which are highly active tyrosine kinases that drive abnormal cell proliferation (embodying an Achilles heel). However, Imatinib therapy leads to the appearance of multifactorial drug resistance in a significant proportion of subjects, including the prevalent T315I ‘gatekeeper’ BCR-ABL mutation that is highly resistant to this drug2. Whether preexisting or induced, the scourge of clinical relapse due to on (and off)- target effects of drugs remains a fundamental problem that can now be addressed at all stages of the drug-validation process. A general approach pursued for Imatinib-resistant patients has been to design and validate second generation ABL inhibitors such as Dasatinib and Nilotinib, which target Imatinib-resistant ABL mutants (although not T315I ABL) and new third generation inhibitors such as Ponatinib, a multi-tyrosine kinase inhibitor with activity towards ABL T315I (Table 1, page 50). Although it is too early to know whether novel drug-resistant ABL mutants will evolve after chronic Ponatinibexposure, this drug now forms part of a comprehensive portfolio of ABL inhibitors that will hopefully fulfil their potential and become the panacea that AML and CML patients clearly require[12]. Moreover, once the full potential of deep-sequencing approaches are realised in the field, new kinome sensitivity and resistance phenomena will rapidly be revealed, driving flexible and personalised drug delivery that ensures stable disease management and potentially cure for the great majority of patients.
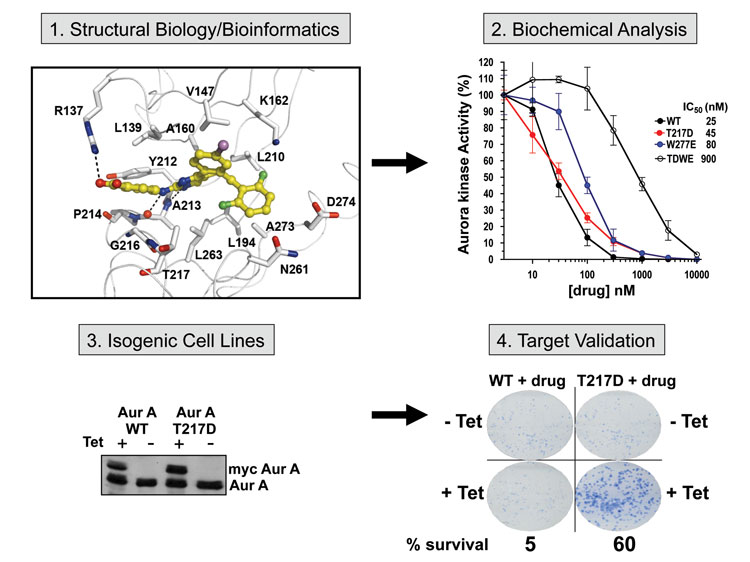
Figure 2 Iterative protocol for predicting, designing, testing and validating drug-resistance alleles. Structural or bioinformatic data is used to investigate likely amino acids involved in binding to kinase inhibitors. These can be tested biochemically and then expressed in Tetracyclin (+Tet) isogenic cancer cell models. Survival of cells in the presence of the compound (quantified by clonogenic assay) provides convincing evidence for an on-target inhibitor effect, with the added benefit of predicting likely drug-resistant mutations that could occur in vivo. Drug-resistant cells can also be subjected to chemical or siRNA screens to identify other molecular vulnerabilities as second-line therapies
Clinical kinase inhibitors inevitably induce drug-resistance in vivo
The first cohort of approved kinase inhibitors target a limited number of tyrosine kinases[3]. Unfortunately, therapy-induced drug resistance is associated with all these protein kinases. However, data from these patients has proved invaluable for revealing patterns of resistance in these kinases that are induced using current drug and delivery combination protocols. For example, tyrosine kinases such as the Epidermal Growth Factor Receptor (EGFR) represent appealing drug targets for the design of small molecule inhibitors, given the high levels of EGFR expression evident in lung cancers. Early investment bore fruit when quinazoline-based inhibitors of EGFR (Gefitinib in 2003, Erlotinib in 2005) were approved for advanced non-small cell lung cancer. Interestingly, patient response improves markedly if tumours are pre-screened for specific oncogenic mutations such as L858R prior to therapy[13], an early example of personalised medicine in this field. Unfortunately, drug resistance is a considerable problem in these patients and the prevalent T790M ‘gatekeeper’ mutation that appears in EGFR induces resistance to currently approved drugs[14]. A similar scenario applies for the small percentage of patients (approximately four percent) whose non-small cell lung cancer is reliant upon an activated EML4-ALK kinase. Pre-selection of patients with the appropriate genotype prior to administration of the approved ALK inhibitor Crizotinib leads to responses, which are often blunted due to ALK drug-resistance at specific positions in the ATP binding site. Following in the footsteps of the Imatinib model, second-generation ALK inhibitors towards such drug-resistant ALK mutants have already been developed[15]. Finally, prevalent drug-resistance (including gatekeeper mutations) has recently been characterised in relapsed patients treated with Vemurafenib, the first of the Ser/Thr BRAF V600E kinase inhibitors approved for malignant melanoma[16]. Together, these findings confirm drug resistance as a kinome-wide issue in cancer biology, and as discussed below, argue for investment in early intervention strategies to help attack this problem. The mechanism of resistance to all these drugs is of central interest. Fortuitously, it appears that a limited number of mutations at key loci account for the vast majority of mutations found in protein kinases (Table 2). The key mutations in protein kinases that appear in patients exposed to kinase inhibitors all have a similar net effect, in that they change the affinity of the kinase for the drug, either directly, by inducing direct steric blockade, or indirectly by activating the kinase or increasing the affinity for ATP[17,18].
Unfortunately, tumours can also become resistant to kinase inhibitors through other mechanisms, including changes in drug influx, efflux and metabolism, modulation of the expression levels of signalling molecules and via drug-induced molecular rewiring of signalling pathways. Excitingly, most of these responses are still potentially druggable, and although research in this area is at an early stage, by optimising different combinations of therapy (including, but not limited to, kinase inhibitors) mechanisms to decrease, prevent or reverse the appearance of drug resistance should be explored.
Kinome-wide drug-resistance towards inhibitors is a predictable problem with rules
All major kinase inhibitors approved for therapy elicit drug-resistance responses in human cells. Mechanistically, this can involve target overexpression, the selection of pre-existing populations of tumour cells with a selective (oncogenic) advantage or the appearance of drug-resistant cells containing a kinase mutation in the highly plastic ATP-binding site. At face value, it might appear that because so many different amino acids variations are encoded in drug-binding sites kinome-wide (indeed, this is the molecular basis for drug specificity), that each inhibitor / kinase combination might contain a unique resistance signature. Fortunately, this is not the case, and the predictable mutational spectrum that appears in all protein kinases targeted in disease, combined with new approaches that emulate the rapid development, testing and approval of drugs like Ponatinib, advocate new models based on experimentally-evaluated drug-resistance and its flexible application to the needs of specific patients. These include chemical genetic approaches for evaluating drug-resistance in cancer cells (see below) and the exploitation of model experimental organisms for probing drugresistance mech anisms such as bacteria[19], yeast[20] and mouse models[16].
Table 2: Major protein kinase motifs associated with kinome-wide drug resistance, with particular emphasis on examples discussed in this review
Name/position of motif mutated | Examples of Drug-Resistant Kinases | Examples of Inhibitors involved |
Gly-rich loop (Gly-X-Gly-X-X-Gly) | ABL, ALK | Imatinib Crizotinib |
Gatekeeper Residue | ABL, EGFR, PDGFR, ALK, BRAF | Imatinib Dasatinib Nilotinib Crizotinib Vemurafenib Gefitinib Erlotinib |
Hydrophobic (Gatekeeper +2) | ALK, AURA, AURB | Crizotinib, VX680 |
Specificity surface (Gatekeeper +6/+7) | ABL, AURA, AURB, PLK1, PLK4 | Imatinib, VX680, MLN8237, BI2536 |
Activation Loop | ABL | Imatinib |
Drug resistance is commonly elicited at three major drug-resistance hotspots that are encoded in all kinase catalytic domains. These are the Gly-rich loop (or P-loop), which clamps and positions the phosphates of ATP, four amino acids that line the ATP-binding site (which we term the ‘resistance tetrad’, Figure 1, page 50) and the T-loop (or activation loop), which regulates the catalytic activity of most kinases[21]. Mutations in the P-loop are common in patients exposed to first-generation kinase inhibitors such as Imatinib, suggesting that there is some flexibility for the specification of novel amino acid side chains that support signalling through an energetically acceptable interaction with ATP, which is unacceptable for drug binding. The second locus for induction of resistance are amino acids in the ATP binding site composed of the ‘gatekeeper’ residue (e.g. T315 of ABL discussed above), a hydrophobic residue positioned two amino acids C-terminal to the gatekeeper, and the proceeding amino acids located in the loop extending from the hinge region, termed the ‘hinge loop’. Figure 1 (page 50) illustrates this ‘resistance tetrad’ in the human kinases ABL and Aurora B and the respective relationship of these amino acids to the molecular surface of the kinase inhibitors Nilotinib and VX680. In concert, these amino acids form a three-dimensional network defining the shape, volume, charge and hydrophobicity of the nucleotide-binding site, which in turn dictates kinase sensitivity/resistance to multiple classes of kinase inhibitor. Since the resistance tetrad represents a very commonly mutated region in patients exposed to small molecule inhibitors, this information has been exploited to create a basic set of mutational ‘rules’ governing how kinases can readily be converted into drug-resistant forms[18,22-25].
Harnessing drug-resistance as a chemical genetic tool for kinase inhibitor validation
One strategic method for extending in vivo drug resistance data, which usually emerges only after late-stage clinical trials, is to model how drug resistance occurs, or can be induced to occur, experimentally. The simplest way to do this is to examine resistance profiles for known drug / kinome groupings, and use this information to model and test how drug resistance can occur in new kinase / inhibitor pairings. We employ an iterative approach combining structural biology, modelling and bioinformatics with biochemistry and cell biology (Figure 2), which can predict and confirm drug-resistance in essentially any inhibitor and protein kinase pairing. For example, kinase inhibitors such as VX680 (Tozasertib) are important pharmacological tools with potential clinical roles. Already established as a pan-Aurora kinase inhibitor[26], VX680 is also a potent ABL inhibitor that by virtue of its binding-mode retains inhibition of the T315I ABL mutant[27]. Using chemical genetic approaches, we found that distinct mutations in the Aurora A or B resistance tetrad (Figure 1) reverse the phenotypic and cytotoxic effects of VX680 in cancer cells, with cell killing requiring specific binding of the drug to Aurora B25. Similar cellular drug resistance models were developed to probe ‘on-target’ effects of the phase III Aurora A inhibitor MLN8237 (Alisertib), through the expression of T217D/E Aurora A mutants[28]. Distinct drug targets such as Polo-like kinase (PLK1) have also been investigated, and an R136G mutation in the PLK1 resistance tetrad induces phenotypic resistance to the preclinical inhibitor BI253625. Interestingly, random mutagenesis promoted by prolonged exposure of genetically unstable cancer cells to BI2536 also induces R136G PLK1, and this mutation is sufficient for cell proliferation in the presence of BI2536[29]. A reverse ‘target-hopping’ approach has also been employed to predict that the resistance tetrad amino acid sequence in the centriole regulating kinase PLK4 might make it vulnerable to Aurora kinase inhibitors such as VX680, and we were able to validate this compound as a sub-micromolar inhibitor of PLK4, binding occurring through a specific inter action validated using a G95R drug resistant mutant[28]. Similar predictive approaches have also been used to discover that p38 MAPK inhibitor can be used as surrogate TGFβ receptor[23] or Casein Kinase 1[30] inhibitors.
Conclusion
As kinase inhibitors become more entrenched in clinical portfolios worldwide, there are new opportunities to catalogue drug-resistance mutations in a more thorough fashion, informing rapid screening opportunities for next generation compounds[24]. In the case of ABL kinase, this has already led to the development of second and third generation therapies that can override known drug resistance, culminating with the rapid approval of Ponatinib in December 2012. By employing similar tactics, maximum use should be made of the vast numbers of compounds available to both research and clinical communities, ensuring that experimental data pertaining to the sensitivity and resistance of kinases to inhibitors can be rapidly exploited as and when it is needed. Mechanistic evaluation of kinase inhibitor specificity/resistance across the kinome lies at the heart of a scientific approach to coordinating a flexible response to the clinical problem of drug resistance. As a bonus, such studies provide important confidence indicators for the employment of kinase inhibitors in experimental biology, where high specificity is of particular importance. The availability of harmonised, publically available, information pertaining to both common and unique resistance mechanisms elicited by kinase inhibitors across the kinome will be an important next step. These date can then be employed to mount an attack on drug resistance, either through early drug discovery efforts in which known mutated kinases are employed during inhibitor screening and optimisation, or by helping to refine drug combination, delivery or stratification of patients that receive kinase inhibitors. Such approaches include the timing of drug delivery, and might include appropriate ‘drug-holidays’. The latter approach is supported by very recent findings demonstrating that the burden of resistance to the BRAF inhibitor Vemurafenib can be reduced in vivo by discontinuous drug therapy, whereby the ‘addiction’ of melanoma cells to Vemurafenib is exploited by enforcing a period of drug withdrawal in order to eradicate them[31]. Ultimately, the test-bed for all clinical kinase inhibitors are genetically diverse human populations, and in the future we must ensure that clinically-approved kinase inhibitor strategies are securely backed up by an informed plan to counter drug-resistance when, as appears inevitable, it occurs.
References
- Druker, B.J., et al., Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine, 2001. 344(14): p. 1031-7
- Gorre, M.E., et al., Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science, 2001. 293(5531): p. 876-80
- Cohen, P. and D.R. Alessi, Kinase Drug Discovery – What’s Next in the Field? ACS chemical biology, 2012
- Davis, M.I., et al., Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology, 2011. 29(11): p. 1046-51
- Davies, S.P., et al., Specificity and mechanism of action of some commonly used protein kinase inhibitors. The Biochemical journal, 2000. 351(Pt 1): p. 95-105
- Manning, G., et al., The protein kinase complement of the human genome. Science, 2002. 298(5600): p. 1912-34
- Knight, Z.A., H. Lin, and K.M. Shokat, Targeting the cancer kinome through polypharmacology. Nature reviews. Cancer, 2010. 10(2): p. 130-7
- Anastassiadis, T., et al., Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature biotechnology, 2011. 29(11): p. 1039-45
- Patricelli, M.P., et al., In situ kinase profiling reveals functionally relevant properties of native kinases. Chemistry & biology, 2011. 18(6): p. 699-710
- Workman, P. and I. Collins, Probing the probes: fitness factors for small molecule tools. Chemistry & biology, 2010. 17(6): p. 561-77
- Cohen, P., Guidelines for the effective use of chemical inhibitors of protein function to understand their roles in cell regulation. The Biochemical journal, 2010. 425(1): p. 53-4
- Cortes, J.E., et al., Ponatinib in refractory Philadelphia chromosome-positive leukemias. The New England journal of medicine, 2012. 367(22): p. 2075-88.
- Paez, J.G., et al., EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 2004. 304(5676): p. 1497-500
- Pao, W., et al., Acquired resistance of lung adeno – carcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine, 2005. 2(3): p. e73
- Katayama, R., et al., Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine, 2012. 4(120): p. 120ra17
- Whittaker, S., et al., Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Science translational medicine, 2010. 2(35): p. 35ra41
- Yun, C.H., et al., The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proceedings of the National Academy of Sciences of the United States of America, 2008. 105(6): p. 2070-5
- Balzano, D., et al., A general framework for inhibitor resistance in protein kinases. Chemistry & biology, 2011. 18(8): p. 966-75
- Azam, M., R.R. Latek, and G.Q. Daley, Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell, 2003. 112(6): p. 831-43
- Kawashima, S.A., et al., Analyzing fission yeast multidrug resistance mechanisms to develop a genetically tractable model system for chemical biology. Chemistry & biology, 2012. 19(7): p. 893-901
- Krishnamurty, R. and D.J. Maly, Biochemical mechanisms of resistance to small-molecule protein kinase inhibitors. ACS chemical biology, 2010. 5(1): p. 121-38
- Barouch-Bentov, R. and K. Sauer, Mechanisms of drug resistance in kinases. Expert opinion on investigational drugs, 2011. 20(2): p. 153-208
- Eyers, P.A., et al., Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chemistry & biology, 1998. 5(6): p. 321-8
- Carter, T.A., et al., Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(31): p. 11011-6
- Scutt, P.J., et al., Discovery and exploitation of inhibitor-resistant aurora and polo kinase mutants for the analysis of mitotic networks. The Journal of biological chemistry, 2009. 284(23): p. 15880-93
- Tyler, R.K., et al., VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell cycle, 2007. 6(22): p. 2846-54
- Young, M.A., et al., Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer research, 2006. 66(2): p. 1007-14
- Sloane, D.A., et al., Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS chemical biology, 2010. 5(6): p. 563-76
- Wacker, S.A., et al., Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nature chemical biology, 2012. 8(3): p. 235-7.
- Verkaar, F., et al., Inhibition of Wnt/beta-catenin signaling by p38 MAP kinase inhibitors is explained by cross-reactivity with casein kinase Idelta/varepsilon. Chemistry & biology, 2011. 18(4): p. 485-94
- Das Thackur, M., et al., Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. In Press Nature doi:10.1038/nature11814
Biographies
 Patrick A. Eyers graduated in Biochemistry from the University of Bristol in 1997 and completed his PhD in 2000 with Sir Philip Cohen in the MRC Protein Phosphorylation Unit at the University of Dundee, eventually swapping the Speedwell Bar for a postdoctoral position with Jim Maller in Denver, Colorado, where he helped establish the Recovery Room Bar Soccer Team (bar and team since disbanded for tax evasion). As an HHMI postdoctoral fellow, he investigated how protein kinases regulate the cell cycle in the model Xenopus laevis oocyte system. His research lab was established at the University of Manchester in 2006, with funding from an MRC Career Development Fellowship and CR-UK. He is currently Senior Lecturer in the Department of Oncology at the University of Sheffield and a member of the Sheffield Cancer Research Centre.
Patrick A. Eyers graduated in Biochemistry from the University of Bristol in 1997 and completed his PhD in 2000 with Sir Philip Cohen in the MRC Protein Phosphorylation Unit at the University of Dundee, eventually swapping the Speedwell Bar for a postdoctoral position with Jim Maller in Denver, Colorado, where he helped establish the Recovery Room Bar Soccer Team (bar and team since disbanded for tax evasion). As an HHMI postdoctoral fellow, he investigated how protein kinases regulate the cell cycle in the model Xenopus laevis oocyte system. His research lab was established at the University of Manchester in 2006, with funding from an MRC Career Development Fellowship and CR-UK. He is currently Senior Lecturer in the Department of Oncology at the University of Sheffield and a member of the Sheffield Cancer Research Centre.
