Chloride ion channels and transporters: from curiosities of nature and source of human disease to drug targets
Posted: 20 August 2013 | | No comments yet
Early in their undergraduate education, the student is introduced to various types of integral membrane protein: receptors, adhesion proteins, ion channels, ion pumps and ion transporters. As they progress through their studies, they find out that discrete gene families and protein structures are responsible for these different protein classes and there is never any reason to consider that there might be any ambiguity in assigning any particular protein to its appropriate protein class.
This satisfies virtually every case from the classroom to the research lab until we consider chloride channels. We know that they exist, are important and might make good drug targets in several disease areas, but until recently, they have remained somewhat stigmatised and unfashionable in the world of therapeutics. The lack of selective ligands has not helped at all. The only exception to this has been the GABA-A receptor that is coupled to an intrinsic chloride channel that opens upon binding of the inhibitory neurotransmitter GABA. Benzodiazepines have been potentiating this receptor ever since Valium became available in the 1960s, providing sedative and anticonvulsant effects. This introduces an important concept: there are several gene families and different protein types that can be described as being chloride channels.
This is in contrast to what we understand of cation-selective ion channels where there is little flexibility in the protein structure that can form a pore that is selective for potassium, sodium, or calcium; the diversity of these ion channels is brought about by the variations in the protein domains distinct from the pore that influence the opening-closing behaviour. To be fair, there do not seem to be many physiological reasons why an anion channel should exhibit chloride-selectivity as there is little physiological role in the membrane transport of other halides or small anions; any anion channel will by default be a chloride channel from a physiological viewpoint. On the other hand, potassium, sodium and calcium membrane currents each play different fundamental roles and selective membrane permeability to these cations is crucial to cellular function and indeed life itself.
GABA-A receptors are members of the cys-loop pentameric ligand-gated ion channel families, which include several types of excitatory and inhibitory neurotransmitter-gated channels. The cystic fibrosis transmembrane regulator (CFTR) protein is a nucleotide-regulated chloride channel. It is a member of the diverse ATP-binding cassette (ABC) transporter family and is the only one that does not seem to transport any substrate across a membrane, but rather functions as a chloride channel. The CLC family of voltage-gated chloride channels arise from another distinct gene family and have a general structure unique among membrane proteins. This family of proteins have brought about many surprises over the last 30 years and members of this family will be introduced here and also how they might be considered drug targets.
Other exciting developments from the chloride channel field have emerged following the molecular identification of proteins underlying calcium-activated chloride (ClCa) channels. Functionally, we have known about their physiological roles for some time, but recent molecular identification of the channel proteins has allowed development of molecular and pharmacological tools to probe and alter their activity. This review focuses on the pharmacological potential of targeting ClCa channels and CLC proteins; their function and subcellular localisation are summarised in Figure 1.
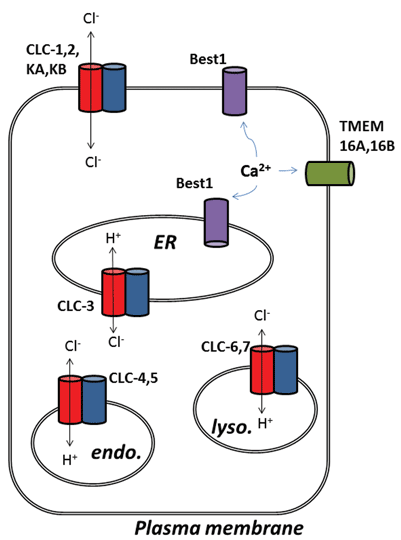
Figure 1
An introduction to CLC proteins
The nine members of this protein family are CLC-1 to 7, CLC-KA and KB. The founder member, CLC-1, is the voltage-gated chloride channel of skeletal muscle and will be discussed below. Many functional studies have been carried out on a homologue with the honorary title of CLC-0, isolated from the Electroplax electric organ, which enables the ray to stun its aquatic victims. Reconstitution of CLC-0 into lipid bilayers and electrophysiological recording of currents flowing through single ion channels revealed an intriguing property. Usually, when recording current from a small patch of membrane containing a single ion channel, one observes fluctuations between two current amplitudes: one representing the closed channel and the other representing the current flowing through the open channel. The fluctuations reflect transitions between open and closed kinetic states. With CLC-0, however, it was evident that the single ion channel protein consisted of two equivalent pores that could open independently1. This channel was therefore described as being double-barrelled (Figure 2). It thus came of no great surprise that the crystal structures of homologues from enteric bacteria revealed that the protein complex was a dimer, with each subunit possessing an ion-conducting pathway2 (Figure 2).
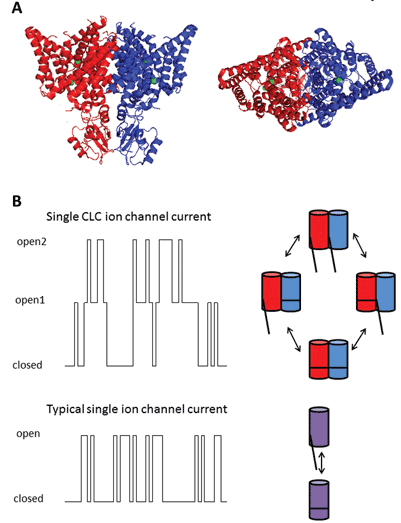
Figure 2
The second surprise arose when detailed functional experiments were carried out on CLC-ec1 from E. coli. Rather than functioning as a chloride channel, like its long-lost relative in vertebrate skeletal muscle, CLC-ec1 is a 2Cl–/H+ exchange transporter, or antiporter, with strict 2:1 exchange stoichiometry3. This protein is important for the survival of enteric bacteria in low pH4 and is likely to exploit a chloride gradient to keep cellular pH at a tolerable level. Prokaryotic CLCs might therefore be targets for inhibitors to treat pathogenic E. coli or Salmonella infection4. The discovery of ion exchange behaviour naturally stimulated further exertions to determine whether any of this chloride-for-proton exchange existed in mammalian CLCs, on the assumption that Cl–/H+ exchange might be the true function of archetypal CLCs and those identified as chloride channels might just be exceptions to the rule. This seems to be the case as there is strong evidence that CLC-3 through to CLC-7 function as 2Cl–/H+ exchange transporters, primarily residing in intracellular organelles, whilst CLC-1, CLC-2, KA and KB are true plasma membrane chloride-conducting ion channels5-9. Because the archetypal CLC property is exchange transport, being found across all forms of cellular life, the latter subclass of proteins, the bona fide chloride channels, can therefore be considered ‘broken’ chloride transporters, which have lost the coupling of chloride transport to the movement of a second substrate ion.
CLC-1
This founder of the CLC family is the voltage-gated chloride channel of skeletal muscle and serves to regulate the membrane potential and repolarise the membrane following action potentials to relax the muscle. In most tissues, we would expect potassium channels to play this role, which they do in cardiac and smooth muscle, as well as regulating membrane excitability in other cell types. The transverse tubule system is an extension to the plasma membrane (sarcolemma) and penetrates into the contractile tissue. It is critical for the rapid spread of electrical activity throughout the muscle and orchestrates rapid and controlled contraction. Potassium efflux from the muscle cells into the confined space of the t-tubule would raise the extracellular potassium concentration and collapse this ion gradient, which would lead to prolonged membrane depolarisation. With chloride channels playing the predominant repolarising role, this is prevented. Loss of function mutations in CLC-1 lead to myotonia in man, goats, and mice (for a recent review see10) and is characterised by impaired muscle relaxation, consistent with the loss of a repolarising membrane current.
With expression confined to skeletal muscle, CLC-1 might be an attractive target for drugs that control muscle contraction by increasing or decreasing CLC-1 function and thereby reduce or increase muscle excitability, respectively. Compounds that increase CLC-1 function might be able to treat myotonia, particularly in cases where they could compensate for partial loss of chloride channel activity. Inhibition of CLC-1 could reduce the threshold for muscle contraction and may be useful in cases of muscle weakness or degenerative diseases such as muscular dystrophy.
CLC-2
The inwardly-rectifying chloride channel, CLC-2, has somewhat widespread tissue distribution. It can be found in central neurones where it regulates neuronal activity11-13. In astrocytes, its subcellular targeting to cell junctions is regulated by an interaction with GlialCAM (MLC1), mutations in which disrupt this targeting and cause megalencephalic leukoencephalopathy14. Recent therapeutic interests involve CLC-2 expression in gut mucosa and lung bronchioles where it plays a role in intestinal and lung secretions, respectively. Lubiprostone, a drug used clinically to relieve constipation was proposed to exert its effect by activating CLC-2, but this effect is controversial. Whilst this compound activates CLC-2 channels in some studies, in others it regulates CLC-2 trafficking and increases CFTR function via prostaglandin receptor activation15,16. However, there remains sufficient evidence that increasing airway CLC-2 function could provide an alternative chloride pathway in cystic fibrosis17.
CLC-KA/KB
These chloride channels are notably expressed in renal epithelia and contribute to the permeability of cell membranes to passive chloride flux. They have a key role in the ascending limb and distal tubules of the nephron, providing a basolateral route for chloride reabsorption, following transport from the primary urine via the apical membrane. The CLCKB gene, which encodes CLC-KB, is one of five genes that underlie Bartter’s syndrome. Loss-of-function mutations in CLC-KB result in defective chloride reabsorption and so leads to a salt-wasting disorder, which is associated with polyuria. One of the main features of individuals that are affected by CLC-KB mutations (Bartter’s type III) is low blood pressure. A more severe form of the disease is caused by mutations in the BSND gene (Bartter’s type IV), which encodes Barttin, an accessory protein important for the trafficking of both CLC-KA and KB to the plasma membrane18. This form of the disorder also includes sensory deafness, which is thought to be brought about by the loss of trafficking of both CLC-KA and KB to epithelial membranes in inner ear. Presumably, loss of either CLC-KA or KB activity, but not both, can be tolerated by the auditory system. This suggests that inhibitors selective for either CLC-KA or CLC-KB, or partial inhibition of both, may act as a novel loop diuretic with the potential to lower blood pressure and with few side-effects. This has led to the study of their pharmacology and the development of novel inhibitor derivatives with low micromolar affinity19, which had diuretic effects when administered to rats20. On the other hand, drugs that activate CLC-KB channels may enhance residual activity of defective channels in Bartter’s type III patients.
CLC-7
Of the 2Cl–/H+ exchange transporter subclass, CLC-7 is a promising target for drug action and its inhibition may be beneficial in osteoporosis. Once more, this indication takes its origin from observations of human disease caused by loss-of-function mutations in CLC-7, which cause osteropetrosis21. In this disorder, bone remodelling by osteoclasts is deficient, which is thought to be caused by defective acid and enzyme secretion. This leads to dense bone, which if reproduced by a CLC-7 inhibitor could reduce the dissolution of bone and therefore strengthen the skeleton of osteoporosis patients22,23. In proof-of-concept studies, pharmacological inhibition of acidification24 or disruption of CLC-7 function by antibodies25 reduced bone resorption. However, CLC-7 inhibition may not be without complications because human and mouse studies suggest that lack of CLC-7 function may also be associated with neuronal storage and degeneration disorders because of reduced lysosomal function26.
Loss-of-function human mutations – lessons from CFTR
It is worth a brief pause at this point to consider that some of the therapeutic indications for novel CLC-targeted drugs aim to treat disorders distinct and at the opposite end of the spectrum to those that are caused by defective CLC function. In addition to those described above, Dent’s I disease is an X-linked kidney disease that is caused by loss of CLC-5 function (see27 for a recent review). All are rare inherited disorders and a major protein defect involves either reduced protein activity or trafficking to the target membrane. In fact, many individual mutations cause ER-retention and a lack of protein maturation. A parallel could be made with cystic fibrosis, with most of the affected individuals possessing the ER-retained ΔF508 mutation. Recent and stratified approaches to treating cystic fibrosis have been the result of a dual-pronged attack to correct mutant protein folding (CF correctors) and/or increase the activity of plasma membrane CFTR chloride channels (CF potentiators). Success is emerging with the potentiator class28, which are effective in patients with mutations that reduce CFTR activity without loss of protein biosynthesis or trafficking (e.g. G551D), however the effectiveness of folding correctors (which is required in the majority of cases) remains to be established29. Although the diseases associated with loss of CLC function are all rare genetic disorders, we may one day be able to treat individuals with myotonia, Bartter’s syndrome, Dent’s disease and osteopetrosis, as well as cystic fibrosis with drugs that correct the actual cause of their disorder.
Calcium-activated chloride channels
Finally, mention will be made of calcium-activated chloride (ClCa) channels, which have clearly-defined physiological roles in a number of cell types, yet their molecular identification suffered a few false and stuttered starts. The story involves four types of protein: members of CLCA, Bestrophin, Tweety and TMEM16 gene families. In all cases, their recombinant overexpression resulted in the generation of membrane chloride currents that are stimulated by raising the intracellular calcium concentration and, to varying extents, membrane depolarisation. The journey for CLCA came to an end when it was found that this was a protein that was secreted, but also likely upregulated the membrane expression of ClCa channels endogenous to the expression system30. Tweety and Bestrophin proteins do not exhibit all of the properties of ClCa channels studied in major tissues, although Best1 appears responsible for a component of ClCa in sensory neurons31 and may also be an important regulator of calcium release from the endoplasmic reticulum32,33. Three independent studies proposed TMEM16A (also called Ano1) as a candidate for a major component, if not in its entirety, for a calcium-activated chloride channel34-36. Many subsequent studies, facilitated by the generation of molecular tools, have supported this. TMEM16A is important for regulating membrane excitability in vascular smooth muscle, is upregulated in an animal model of pulmonary hypertension and tone can be reduced by channel inhibition37,38. In sensory neurones, TMEM16A couples the presence of inflammatory mediators to membrane hyperexcitability and TMEM16A inhibition has antinociceptive effects39. In animal models of asthma, expression of this particular chloride channel is increased, and its inhibition may have beneficial effects40. It is also found in interstitial cells of Cajal of the intestine and channel function is required for the rhythmical contraction of smooth muscle in the intestinal wall41. Furthermore, TMEM16A activation may provide an alternative pathway for epithelial chloride secretion in cystic fibrosis42. Whilst there is still some debate surrounding the precise function of the rest of the TMEM16 family, ClCa function has been also been ascribed to TMEM16B (Ano2), which is thought to underlie the ClCa channel in olfactory hair cells43,44. There may also be roles for this class of ion channel in cancer cell biology and its inhibition may prevent cell proliferation45-48.
Concluding remarks
This review has highlighted the diverse roles of chloride conducting or transporting proteins and how their dysfunction is coupled to human disorder or disease-like symptoms in animal models. There is a severe lack of pharmacological reagents that inhibit, activate or improve membrane trafficking of chloride channels and transporters. Progress is being made with drugs that reverse the defective function of CFTR in cystic fibrosis, which will hopefully result in medications that are specific to the different types of inherited mutation. Inhibitors and activators of TMEM16A calcium-activated chloride channels42,49 are proving useful laboratory tools and new compounds might make effective drugs, particularly if they can deliver tissue-specific effects. Molecules specific for particular CLCs are probably the most elusive. Understanding the structural basis of voltage-gated CLC activation50 might identify protein domains that are targetable by rational structure-based drug design. Such tools will enable us to put some of the novel therapeutic ideas that have been reviewed and presented here to the test.
References
- Miller, C., Open-state substructure of single chloride channels from Torpedo electroplax. Philos Trans R Soc Lond B Biol Sci, 1982. 299(1097): p. 401-11
- Dutzler, R., et al., X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature, 2002. 415(6869): p. 287-94
- Accardi, A. and C. Miller, Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature, 2004. 427(6977): p. 803-7
- Iyer, R., et al., A biological role for prokaryotic ClC chloride channels. Nature, 2002. 419(6908): p. 715-8
- Scheel, O., et al., Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature, 2005. 436(7049): p. 424-7
- Picollo, A. and M. Pusch, Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature, 2005. 436(7049): p. 420-3
- Leisle, L., et al., ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity. EMBO J, 2011. 30(11): p. 2140-52
- Neagoe, I., et al., The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J Biol Chem, 2010. 285(28): p. 21689-97
- Matsuda, J.J., et al., Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol, 2008. 294(1): p. C251-62
- Tang, C.Y. and T.Y. Chen, Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia. J Biomed Biotechnol, 2011. 2011: p. 685328
- Ratte, S. and S.A. Prescott, ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J Neurosci, 2011. 31(44): p. 15838-43
- Foldy, C., et al., Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat Neurosci, 2010. 13(9): p. 1047-9
- Rinke, I., J. Artmann, and V. Stein, ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J Neurosci, 2010. 30(13): p. 4776-86
- Jeworutzki, E., et al., GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(-) channel auxiliary subunit. Neuron, 2012. 73(5): p. 951-61
- Norimatsu, Y., A.R. Moran, and K.D. MacDonald, Lubiprostone activates CFTR, but not ClC-2, via the prostaglandin receptor (EP(4)). Biochem Biophys Res Commun, 2012. 426(3): p. 374-9
- Cuppoletti, J., et al., SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol Cell Physiol, 2004. 287(5): p. C1173-83
- Cuppoletti, J., et al., ClC-2 Cl- channels in human lung epithelia: activation by arachidonic acid, amidation, and acid-activated omeprazole. Am J Physiol Cell Physiol, 2001. 281(1): p. C46-54
- Estevez, R., et al., Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature, 2001. 414(6863): p. 558-61
- Liantonio, A., et al., Molecular switch for CLC-K Cl- channel block/activation: optimal pharmacophoric requirements towards high-affinity ligands. Proc Natl Acad Sci U S A, 2008. 105(4): p. 1369-73
- Liantonio, A., et al., In-vivo administration of CLC-K kidney chloride channels inhibitors increases water diuresis in rats: a new drug target for hypertension? J Hypertens, 2012. 30(1): p. 153-67
- Kornak, U., et al., Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell, 2001. 104(2): p. 205-15
- Schaller, S., et al., The role of chloride channels in osteoclasts: ClC-7 as a target for osteoporosis treatment. Drug News Perspect, 2005. 18(8): p. 489-95
- Zhao, Q., et al., CLC-7: a potential therapeutic target for the treatment of osteoporosis and neurodegeneration. Biochem Biophys Res Commun, 2009. 384(3): p. 277-9
- Karsdal, M.A., et al., Acidification of the osteoclastic resorption compartment provides insight into the coupling of bone formation to bone resorption. Am J Pathol, 2005. 166(2): p. 467-76
- Ohgi, K., et al., Antibodies against ClC7 inhibit extracellular acidification-induced Cl(-) currents and bone resorption activity in mouse osteoclasts. Naunyn Schmiedebergs Arch Pharmacol, 2011. 383(1): p. 79-90
- Kasper, D., et al., Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J, 2005. 24(5): p. 1079-91
- Lippiat, J.D. and A.J. Smith, The CLC-5 2Cl(-)/H(+) exchange transporter in endosomal function and Dent’s disease. Front Physiol, 2012. 3: p. 449
- Ramsey, B.W., et al., A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med, 2011. 365(18): p. 1663-72
- Clancy, J.P., et al., Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax, 2012. 67(1): p. 12-8
- Gibson, A., et al., hCLCA1 and mCLCA3 are secreted non-integral membrane proteins and therefore are not ion channels. J Biol Chem, 2005. 280(29): p. 27205-12
- Boudes, M., et al., Best1 is a gene regulated by nerve injury and required for Ca2+-activated Cl- current expression in axotomized sensory neurons. J Neurosci, 2009. 29(32): p. 10063-71
- Kunzelmann, K., et al., Role of the Ca2+ -activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol Chem, 2011. 392(1-2): p. 125-34
- Barro-Soria, R., et al., ER-localized bestrophin 1 activates Ca2+-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Arch, 2010. 459(3): p. 485-97
- Schroeder, B.C., et al., Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell, 2008. 134(6): p. 1019-29
- Caputo, A., et al., TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science, 2008. 322(5901): p. 590-4
- Yang, Y.D., et al., TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature, 2008. 455(7217): p. 1210-5
- Davis, A.J., et al., Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) -A01. Br J Pharmacol, 2013. 168(3): p. 773-84
- Forrest, A.S., et al., Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am J Physiol Cell Physiol, 2012. 303(12): p. C1229-43
- Liu, B., et al., The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J Clin Invest, 2010. 120(4): p. 1240-52
- Zhang, C.H., et al., The TMEM16A Ca2+-activated Cl- Channel in Airway Smooth Muscle Contributes to Airway Hyperresponsiveness. Am J Respir Crit Care Med, 2012
- Hwang, S.J., et al., Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol, 2009. 587(Pt 20): p. 4887-904
- Namkung, W., et al., Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J, 2011. 25(11): p. 4048-62
- Stephan, A.B., et al., ANO2 is the cilial calcium-activated chloride channel that may mediate olfactory amplification. Proc Natl Acad Sci U S A, 2009. 106(28): p. 11776-81
- Hengl, T., et al., Molecular components of signal amplification in olfactory sensory cilia. Proc Natl Acad Sci U S A, 2010. 107(13): p. 6052-7
- Carneiro, A., et al., Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC Cancer, 2008. 8: p. 98
- Stanich, J.E., et al., Ano1 as a regulator of proliferation. Am J Physiol Gastrointest Liver Physiol, 2011. 301(6): p. G1044-51
- Mazzone, A., et al., Inhibition of cell proliferation by a selective inhibitor of the Ca(2+)-activated Cl(-) channel, Ano1. Biochem Biophys Res Commun, 2012. 427(2): p. 248-53
- Duvvuri, U., et al., TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res, 2012. 72(13): p. 3270-81
- Namkung, W., P.W. Phuan, and A.S. Verkman, TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem, 2011. 286(3): p. 2365-74
- Smith, A.J. and J.D. Lippiat, Voltage-dependent charge movement associated with activation of the CLC-5 2Cl-/1H+ exchanger. FASEB J, 2010. 24(10): p. 3696-705
- Feng, L., et al., Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science, 2010. 330(6004): p. 635-41
Author Biography

Dr Jon Lippiat completed his PhD on the structure, function and pharmacology of potassium channels at the University of Leicester. He studied pancreatic beta-cell function and diabetes at the University of Oxford before his appointment as Lecturer in Pharmacology at the University of Leeds. His research involves elucidating the structural and physiological properties of several different types of ion channel and transporter and their potential targeting by novel pharmacological reagents.