

Cell-based assays for protein-protein interactions
Posted: 22 October 2013 | | No comments yet
Protein-protein interactions (PPI) form the backbone of all cellular signalling networks, and aberrant PPI contribute to the pathology of several diseases. Thus, strategies to identify PPI modulators are expected to be therapeutically beneficial. However, there are very few examples of clinically approved PPI modulators, reflecting the difficulties of identifying effective compounds for this target class. This perspective reviews the challenges associated with targeting PPI, and summarises the major strategies used to detect and disrupt PPI, with a particular focus on cell-based assays for PPI.
‘Conventional’ targets versus PPI
To date, enzymes and G protein-coupled receptors (GPCRs) are the protein classes on which most drug discovery efforts have been focused. In addition to the sine qua non of target validation, several other factors ensured that these proteins commanded the attention of pharmaceutical companies. Enzymes have well-defined and relatively ‘compact’ catalytic pockets, making them amenable to inhibition with small molecules. Additionally, enzymatic activity facilitates the design of in vitro biochemical assays for small molecule antagonists. Similarly, based on the discovery of binding pockets for the small endogenous ligands on GPCRs, many small molecule modulators of receptor activity have been identified. In both cases, this greatly aids the iterative structure-function analysis process as medicinal chemists seek to improve both potency and selectivity / specificity for the target.
The huge emphasis on small molecule antagonists in these areas, combined with intense competition for market share between companies, means other disease-relevant target classes have perhaps been neglected. Among these, it is clear that PPI represent an opportunity for drug discovery. For example, in cancer, the oncogenic effects of the MYC and NOTCH transcription factors and the MDM2 and WNT oncogenes are mediated via PPI1-4. Furthermore, current estimates suggest there are between 130,000 and 600,000 human PPI compared to ~22,000 individual proteins, offering the potential for very selective targeting of signalling nodes in diverse pathways5,6.
Chemical modulation of PPI
There are significant challenges associated with targeting PPI. Typically, the interface of a PPI is an extended, rather flat surface with several shallow binding sites, which is thermodynamically unfavourable for binding of small molecule antagonists. Therefore many of the current compound libraries built around classical targets may not contain the required chemical structure for effective PPI antagonism. This has been addressed by the design of additional libraries that contain fragments, peptides, or macrocycles from natural and synthetic sources. In fragment-based screens, several low molecular weight (<200 Da) compounds that bind distinct sites on a protein surface are identified7. Ultimately, chemical stitching of optimised derivatives of the initial fragments can give rise to compounds capable of disrupting PPI. Notable examples of PPI antagonists that were optimised via fragment-based approaches include ABT-737 and SP4206. ABT-737 unleashes the apoptotic activities of the BAK/BAX proteins by releasing them from inhibitory complexes containing anti-apoptotic BCL2 family members8. SP4206 is an antagonist of the interaction between the IL2 cytokine and its receptor9. Both these molecules performed excellently in preclinical studies, and ABT-263, an orally bioavailable derivative of ABT-737, has progressed into several clinical trials10.
PPI can also be disrupted using peptide mimetics, which are based on a contiguous epitope at the PPI interface surface of one of the binding partners. Various amino acid substitutions and modifications to the peptide are often performed to preserve an ‘active’ conformation, aid solubility and confer resistance to degradation. Examples include the so-called ‘stapled’ alpha helix peptides pioneered by Verdine and colleagues11. The backbones of these peptides are chemically modified with a hydrocarbon chain that locks the peptide in its alpha-helical configuration. This procedure provides stability to the molecule and increases the amount of functional peptide compared to non-modified chains. Cancer-relevant PPI that have been successfully targeted include BAX/BCL2, P53/MDM2/4 and NOTCH1/MAML112–14. Backed by both Pharma and venture capital, at least one stapled peptide has progressed into Phase I clinical trials15. However, the target in this case is an extracellular receptor and not an intracellular PPI. Thus, whether intracellular PPI can be successfully targeted in the clinic using stapled peptides remains to be seen.
Similar to the stapling approach, macrocycling aims to stabilise the biologically active conformation of a peptide segment. Again, most advanced projects are focused on modulation of extracellular targets. However, nature has already used this process to cyclise a polypeptide capable of modulating intracellular PPI16. Playing evolutionary catch-up, several pharmaceutical companies are investing in an array of technologies in order to accelerate discovery of therapeutic macrocycles capable of hitting intracellular targets17. Although ‘PPI modulation’ is often conceived as ‘PPI inhibition’, there are examples where preservation of a PPI may be desirable18. Notably, natural macrocycles can either stabilise or inhibit PPI19,20. Thus, structure-guided design may yield synthetic macrocycles that elicit either outcome.
Assays for PPI
In addition to the selection of compound libraries, careful choice of assay format is also important for interrogation of PPI. In vitro screens using purified proteins have clear advantages in terms of defining structure-activity relationships. The benefits of in vitro PPI screens have been thoroughly discussed21. However, there is an obvious lack of cellular context in an in vitro approach, and thus cell-based screens for PPI have potential advantages as outlined below.
Compounds showing activity against a PPI in cell-based screens are by definition cell-permeable. This provides additional information not available from biochemical assays. Also, some PPI are very specific to subcellular localisation (cytosol versus nucleus, for example), or take place in the context of cellular membranes, where the dynamics of the interaction are likely very different to the situation in vitro22. Furthermore, a cellular context facilitates detection of compounds that modulate PPI in an indirect manner. However, such ‘indirect hits’ pose significant challenges in terms of deconvolution to identify the cellular target.
It is not likely that cell-based PPI assays will entirely replace in vitro PPI screens. Indeed, the timing at which a cell based PPI screen is introduced can depend on both the size of the screen itself, and the complexity of the readout. One potential strategy is to perform a primary screen on larger collections of compounds in vitro, and then to use a cell-based PPI screen as part of the hit confirmation process. Alternatively, a cell-based screen may be introduced from the outset if the compound set is limited. Once the decision to perform a cell-based screen is made, there are several options available, a selection of which is discussed below. For a review of additional PPI approaches in several organisms, the reader is referred to23.
BiFC
A large panel of fluorescent proteins (FPs) have been used for the study of PPI in mammalian cells24. The principle of this so-called bimolecular fluorescence complementation (BiFC) is to fuse the N- and C-terminal fragments of a reporter FP to the interacting partners of interest. Upon PPI, the FP fragments are brought into proximity and refold, thus reconstituting an excitable FP (Figure 1). The C-terminal amino acid sequences of GFP, YFP and BFP are identical after amino acid 155, whereas their N termini differ at several residues. In this way, the interaction of one protein with two potential partners can be interrogated simultaneously25. BiFC also offers some advantages over other fluorescence-based methods for PPI detection. For example, Förster resonance energy transfer (FRET) requires careful control of the stoichiometry between interacting partners and significant signal deconvolution, whereas BiFC is relatively free of these issues26.
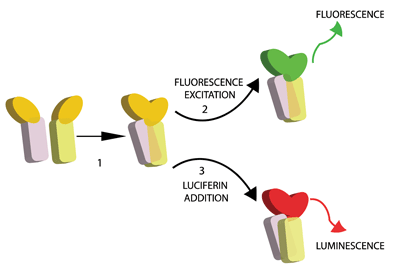
Figure 1: Principle of protein fragment complementation. Two interacting proteins are fused to N and C terminal fragments of a fluorescent or luminescent reporter protein (generic depicted in yellow). Upon interaction, the activity of the reporter is reconstituted and can be read out as fluorescence upon excitation (2) or as luminescence upon substrate addition (3)
Currently, the use of BiFC for discovery of small molecule PPI antagonists is limited to a few examples27,28. The main reason for this is that refolding of many fluorescent chromophore fragments is essentially irreversible29,30. A potential solution to this problem is to engineer an inducible BiFC reporter (using a doxycycline-responsive promoter, for example). Compounds can then be introduced to cells prior to expression of the BiFC constructs. Another issue with BiFC (and other protein fragment complementation assays) is that the signal intensity is often orders of magnitude lower than that obtained with the full-length protein. This reduces the signal:noise ratio, which may compromise assay robustness and thus suitability for HTS. To address these issues, evolution of BiFC partners through mutagenesis will be required. Indeed, several novel FP variants of that enhance signal:noise in BiFC assays have been identified in this manner24. Following re-association of the split chromophore, a maturation step must take place, and in some cases this is temperature-dependent. For example, the signal from some YFP fragments and from mCherry in BiFC is greatly enhanced if the temperature is lowered to 25-30 degrees prior to readout26,31. This presents an additional logistical consideration and potential source of variability that could be associated with BiFC if used in HTS campaigns.
BiLC
In the last few years, the use of split luciferase (BiLC) has emerged as a powerful tool for probing PPIs both in vitro and in vivo32. This technique currently offers several advantages over BiFC. The interaction between the luciferase N and C terminal fragments does not require covalent bond formation, and is therefore completely reversible30. Second, the low affinity of interaction between the unfused split luciferase fragments ensures that the PPI under investigation drives the luminescent output. Additionally, the enzymatic nature of the assays combined with a low background signal provides a robust assay format as measured by screening metrics such as the Z’ score33. PPI involved in cancer biology34,35, Alzheimer’s disease36 and plant signalling37 have all been interrogated using BiLC. Furthermore, BiLC has been used successfully in xenograft studies in mice32,35. Notwithstanding the caveats of the xenograft approach, this opens up the possibility of further characterisation of lead compounds in preclinical studies. BiLC can also be multiplexed. Rational design of complementing click beetle red and click beetle green (CBR and CBG) fragments was performed based on earlier studies of firefly luciferase complementation38. Using ‘bait’ fused to the C terminus of CBG and prey fused to either the N terminus of CBG or CBR, simultaneous detection of two different PPIs was demonstrated38 (Figure 2).
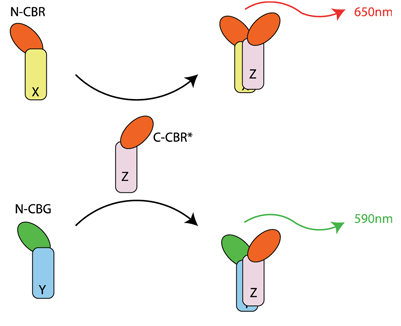
Figure 2: Multiplexing luciferase protein fragment complementation permits simultaneous detection of a protein (Z) interacting with two alternative partners (X or Y). This relies on the principle that the emission spectrum of luciferase is dictated by the N terminal luciferase fragment. Thus, proteins X and Y are fused to the N termini of click beetle red or click beetle green, respectively. They are then interrogated for their ability to interact with a common downstream target fused to a mutated variant of the click beetle red C terminus (CBR*)
BiLC does have limitations, such as sensitivity to false positives (inhibitors of luciferase enzymatic activity, for example) and false negatives (such as auto fluorescent test compounds). Additionally, as with all protein fragment complementation assays, the choice and orientation of fusion partners may be limited to some extent by steric effects. Controls for normalisation of cell number or transfection efficiency are also critical in this assay. Suitable controls include co-transfection or IRES-driven expression of a reporter such as Renilla luciferase or fluorescent protein.
FRET and FLIM
Förster resonance energy transfer (FRET) describes the process whereby energy is transferred non-radiatively from one excited donor FP to an acceptor molecule, which is also often an FP. Careful matching of the paired FPs and their fusion to interacting partners can be used to report PPI39. TR-FRET is a modified version in which a kinetic element is introduced into the assay to reduce interference from assay reagents and compound auto fluorescence. In HTS, both approaches are generally used to detect PPI in a purified protein or cell lysate format. Although, ‘true’ cellular context is lost when using these homogenous assays, they have been successfully used in many screens for PPI antagonists. In contrast, published examples of the use of FRET for HTS campaigns to detect intracellular PPIs are rare (for an example, see40). This is likely due to the technical demands associated with development of a robust intracellular FRET assay41,42. Despite the limitations, some success at detecting extracellular, membrane-bound PPI using (TR)FRET in intact cells has been reported43.
Fluorescence lifetime microscopy (FLIM) is emerging as an alternative to measurement of FRET for the study of PPI. Unlike FRET, FLIM is independent of stoichiometry, as it only measures changes in donor fluorescence lifetime following illumination. Specifically, participation of the donor in FRET reactions with the acceptor alters the donor fluorescence lifetime, which can then be used to estimate changes in FRET efficiency in the presence and absence of compounds. However, conventional FLIM is unsuitable for HTS as its measurement is time-consuming and requires sophisticated instrumentation. Recently, FLIM microplate readers compatible with SBS standard lab ware have been introduced on the market (for examples, see44,45). Furthermore, the throughput of FLIM has been improved, to give 96 well plate reads of minutes, rather than hours46.
Perspective
Cell-based PPI assays hold great promise for discovery of novel disease relevant PPI, as well as the chemical probes that modulate them. The development of novel pairs of both FPs and luciferase fragments that are optimised for high signal:noise and reversible interaction studies will accelerate discovery in the field. Furthermore, developing the capability to multiplex the PPI readout with other parameters will be an important milestone. For instance, incorporation of reporter gene activation or high content phenotypic traits, will provide additional information on candidate small molecule ‘hits’ identified during screening. Finally, it should be noted that stabilisation of PPI could also be therapeutically beneficial in some diseases. This understudied area will greatly benefit from innovative approaches to assay development.
Biography

Mark Wade, Center for Genomic Science of IIT@SEMM
Mark Wade completed his PhD in Virology and Molecular Biology at Imperial College London. He subsequently moved to the Salk Institute, California, to work on molecular cell biology of the MYC oncogene and P53/MDM2/MDMX regulatory circuits. In this period he collaborated with both academia and industry on screens for activators of the P53 tumor suppressor. In 2011, he was recruited to the Center for Genomic Science of IIT@SEMM (Milan), one of the 10 research outstations of the Fondazione Istituto Italiano di Tecnologia (IIT) national network. Mark set up and now runs a Screening Unit focused on the development and prosecution of cell-based siRNA and small molecule screens. The unit works closely with other IIT groups and external collaborators on disease-focused projects, with a particular emphasis on oncology.
References
- Kikuchi, A., Kishida, S. & Yamamoto, H. Regulation of Wnt signaling by protein-protein interaction and post-translational modifications. Exp Mol Med 38, 1-10 (2006)
- Dang, C.V. MYC on the path to cancer. Cell 149, 22-35 (2012)
- Shao, H., Huang, Q. & Liu, Z.J. Targeting Notch signaling for cancer therapeutic intervention. Adv. Pharmacol 65, 191-234 (2012)
- Wade, M., Li, Y.C. & Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13, 83-96 (2013)
- Venkatesan, K. et al. An empirical framework for binary interactome mapping. Nat Methods 6, 83-90 (2009)
- Stumpf, M.P. et al. Estimating the size of the human interactome. Proc Natl Acad Sci USA 105, 6959-64 (2008)
- Valkov, E., Sharpe, T., Marsh, M., Greive, S. & Hyvonen, M. Targeting protein-protein interactions and fragment-based drug discovery. Topics Curr Chem 317, 145-79 (2012)
- Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677-81 (2005)
- Wilson, C.G. & Arkin, M.R. Small-molecule inhibitors of IL-2/IL-2R: lessons learned and applied. Curr. Topics Microbiol Immunol 348, 25-59 (2011)
- http://clinicaltrials.gov/ct2/results?term=navitoclax&Search=Search
- Verdine, G.L. & Walensky, L.D. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin. Cancer Res 13, 7264-70 (2007)
- Walensky, L.D. et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466-70 (2004)
- Chang, Y.S. et al. Stapled alpha-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci USA (2013)
- Moellering, R.E. et al. Direct inhibition of the NOTCH transcription factor complex. Nature 462, 182-8 (2009)
- http://www.aileronrx.com/index.php%5D%5Bhttp://clinicaltrials.gov/ct2/results?term=ALRN+-+5281+&Search=Search
- Ke, H. & Huai, Q. Crystal structures of cyclophilin and its partners. Front Biosci 9, 2285-96 (2004)
- Cain, C. Excited about cycling. BIOCENTURY 20, A7–A13 (2012)
- Thiel, P., Kaiser, M. & Ottmann, C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angewandte Chemie 51, 2012-8 (2012)
- Kunicki, J.B. et al. Synthesis and evaluation of biotinylated sansalvamide A analogs and their modulation of Hsp90. Bioorganic Med Chem Lett 21, 4716-9 (2011)
- Choi, J., Chen, J., Schreiber, S.L. & Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239-42 (1996)
- Arkin, M.R., Glicksman, M.A., Fu, H., Havel, J.J. & Du, Y. Inhibition of Protein-Protein Interactions: Non-Cellular Assay Formats. NIH Assay Guidance Manual (2012)
- Grasberger, B., Minton, A.P., DeLisi, C. & Metzger, H. Interaction between proteins localized in membranes. Proc Natl Acad Sci USA 83, 6258-62 (1986)
- Stynen, B., Tournu, H., Tavernier, J. & Van Dijck, P. Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol Mol Biol Rev 76, 331-82 (2012)
- Kodama, Y. & Hu, C.D. Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. BioTechniques 53, 285-98 (2012)
- Hu, C.D. & Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnology 21, 539-45 (2003)
- Kerppola, T.K. Bimolecular Fluorescence Complementation (BiFC) Analysis of Protein Interactions in Live Cells. Cold Spring Harbor Protocols 2013 (2013)
- Amaral, J.D. et al. Live-cell imaging of p53 interactions using a novel Venus-based bimolecular fluorescence complementation system. Biochem Pharmacol 85, 745-52 (2013)
- Zych, C., Domling, A. & Ayyavoo, V. Development of a robust cell-based high-throughput screening assay to identify targets of HIV-1 viral protein R dimerization. Drug Des Dev Ther 7, 403-12 (2013)
- Magliery, T.J. et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. JACS 127, 146-57 (2005)
- Stefan, E. et al. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc Natl Acad Sci USA 104, 16916-21 (2007)
- Fan, J.Y. et al. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem Biophys Res Comm 367, 47-53 (2008)
- Luker, K.E. et al. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA 101, 12288-93 (2004)
- Zhang, J.H., Chung, T.D. & Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67-73 (1999)
- Hida, N. et al. High-sensitivity real-time imaging of dual protein-protein interactions in living subjects using multicolor luciferases. PLoS One 4, e5868 (2009)
- Fan-Minogue, H. et al. Noninvasive molecular imaging of c-Myc activation in living mice. Proc Natl Acad Sci USA 107, 15892-7 (2010)
- Hashimoto, T., Adams, K.W., Fan, Z., McLean, P.J. & Hyman, B.T. Characterization of oligomer formation of amyloid-beta peptide using a split-luciferase complementation assay. J Biol Chem 286, 27081-91 (2011)
- Gehl, C. et al. Quantitative analysis of dynamic protein-protein interactions in planta by a floated-leaf luciferase complementation imaging (FLuCI) assay using binary Gateway vectors. Plant journal 67, 542-53 (2011)
- Villalobos, V. et al. Dual-color click beetle luciferase heteroprotein fragment complementation assays. Chem Biol 17, 1018-29 (2010)
- Day, R.N. & Davidson, M.W. Fluorescent proteins for FRET microscopy: monitoring protein interactions in living cells. BioEssays 34, 341-50 (2012)
- Song, Y., Madahar, V. & Liao, J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann Biomed Eng 39, 1224-34 (2011)
- Jares-Erijman, E.A. & Jovin, T.M. FRET imaging. Nat Biotechnology 21, 1387-95 (2003)
- Zeug, A., Woehler, A., Neher, E. & Ponimaskin, E.G. Quantitative intensity-based FRET approaches–a comparative snapshot. Biophys J 103, 1821-7 (2012)
- Maurel, D. et al. Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal Biochem 329, 253-62 (2004)
- http://www.assaymetrics.com/PlateReaders.htm
- http://www.fluorescenceinnovations.com/LiveCells.html
- Alibhai, D. et al. Automated fluorescence lifetime imaging plate reader and its application to Forster resonant energy transfer readout of Gag protein aggregation. J Biophotonics 6, 398-408 (2013)
