

A continuous and controlled pharmaceutical freeze-drying technology for unit doses
Posted: 2 January 2018 | Jos Corver, Pieter-Jan Van Bockstal, Thomas De Beer | 1 comment
Among the list of over 300 FDA and EMA approved biopharmaceutical products, around 50% are freeze-dried – indicating that freeze-drying is the preferred way of stabilising biopharmaceutical drug products that are unstable in aqueous solution, despite the high cost and long processing time linked to this manufacturing technique.1,2

Freeze drying is a low-temperature drying process, based on the principles of heat and mass transfer, employed to convert aqueous solutions of (heat-)labile materials into solids with sufficient stability for distribution and storage.3 Many biopharmaceuticals have limited stability in aqueous solution and are subject to a number of degradation pathways mediated by water, which might result in a lower potency or even in toxicity of the drug molecule. A pharmaceutical freeze-drying process consists of three consecutive steps:
- During freezing most of the water crystallises to ice, thus concentrating the solutes between the ice crystals. Some of the solutes crystallise during freezing, while those that do not are transformed into a rigid glass
- The ice crystals are removed under vacuum by sublimation (primary drying). Heat is supplied to the frozen product for sublimation, but the product temperature is kept below the collapse temperature to avoid structural product collapse, hence ensuring a solid and rigid dried cake after freeze-drying
- A secondary drying step where most of the unfrozen water (ie, water dissolved in the amorphous phase) is removed by diffusion and desorption.
The most important critical quality attributes evaluated after freeze-drying on randomly selected samples using off-line analytical techniques are: (i) the API state (eg, protein conformation) and stability; (ii) the residual moisture content; (iii) the freeze-dried product cake appearance; (iv) the reconstitution time.
For more than 80 years, pharmaceutical freeze-drying has been performed using an unchanged batch-wise approach, although the handling equipment before (filling) and after (capping and packaging) freeze-drying is continuously operated by nature. A typical pharmaceutical freeze-dryer consists of a vacuum drying chamber in which the pharmaceutical unit doses (vials) are placed on temperature controlled shelves…
Freeze-drying performed via this traditional batch-wise approach has several disadvantages:
- The freezing step is uncontrolled, which has the significant impact on the consecutive drying steps. Freezing initially involves the cooling of all aqueous solutions (vials) in the freeze-dryer until ice nucleation occurs, which is generally below 0°C (ie, supercooling). Ice nucleation is a stochastic event, hence inducing vial-to-vial variation based on the degree of supercooling: a higher degree of supercooling yields a high number of small ice crystals while at a lower degree of supercooling, a lower number of large ice crystals is formed. As a consequence, the size of the ice crystals differs from vial to vial, which affects the individual vial sublimation rate during primary drying4
- Uneven heat transfer in the freeze-drying chamber results in different energy transfers to vials at different locations on the freeze-dryer shelves. For instance, vials situated at the edge of the shelves are exposed to more radiant heat from the warmer surroundings (ie, door and walls of the freeze-dryer) compared to the vials in the middle of the shelves. This vial-to-vial variability in heat transfer results in significant vial-to-vial difference towards product temperature (danger for collapse!) and drying rate.5
Both these disadvantages result in different freeze-drying process conditions in each vial, leading to uncontrolled vial-to-vial and batch-to-batch end product variability. Such a manufacturing approach is in conflict with the recent Quality-by-Design and process analytical technology guidelines from the regulatory authorities, stating that quality should be built into and guaranteed in each dosage form (ie, in each released vial).6-8
- It is a slow, time-consuming and expensive process. The whole cycle may last from one to seven days depending on the product properties and the vial dimensions
- In an industrial environment, thousands of vials are treated per batch, which induces operational risks, such as complicated handling of vials for loading and unloading of the freeze-dryer. Furthermore, since the handling equipment before (filling) and after (capping, packaging) freeze-drying is continuously operated by nature, buffer systems are necessary. This increases the risk of product contamination
- The handling equipment takes up a large area of space, which is very expensive in terms of capital investment and operational costs because of the high standards of cleanliness and sterility
- Different batch freeze-dryer loadings require different process conditions to be optimised and validated
- The installation is subject to various thermal and pressure conditions. This leads to thermal inefficiencies and the transient conditions may not be well defined
- The course of the freeze-drying process cannot be monitored at the scale of the individual vial. The product behaviour in each vial during freeze-drying is unknown
- Upscaling requires complete re-optimisation and re-validation of the process.
An innovative continuous freeze-drying technology for unit doses overcoming all disadvantages and challenges of traditional batch freeze-drying has been developed.9,10
The continuous freeze-drying technology starts with a continuous freezing step where the vials, filled with liquid product, are rapidly rotated along their longitudinal axis (ie, spin-freezing, see Figure 1). The cooling and freezing is achieved by using a flow of sterile gas with a controllable temperature around the rotating vial. Consequently, the resulting frozen product will be spread with a uniform thickness over the entire vial surface (ie, large surface area and thin product layer). The annealing process can be performed by transferring the vials to a chamber with a controlled temperature.
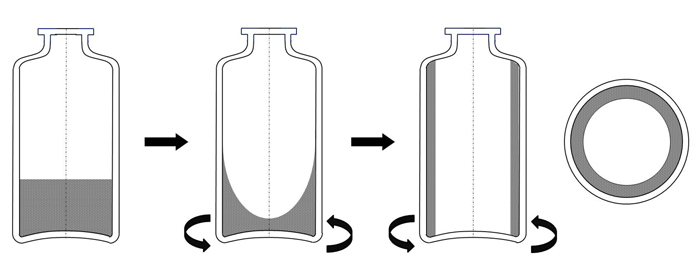
Figure 1: Illustration of the spin freezing step
An appropriate load-lock system is used to rapidly transfer the frozen vials between the continuous freezing and the continuous drying unit, without disturbing the specific conditions of pressure and temperature. In the drying chamber, an endless belt system allows the transport of the vials in front of individually controlled radiators which provide the heat transfer to the vials needed for sublimation and desorption, hence allowing individual vial temperature-regulation (Figure 2).
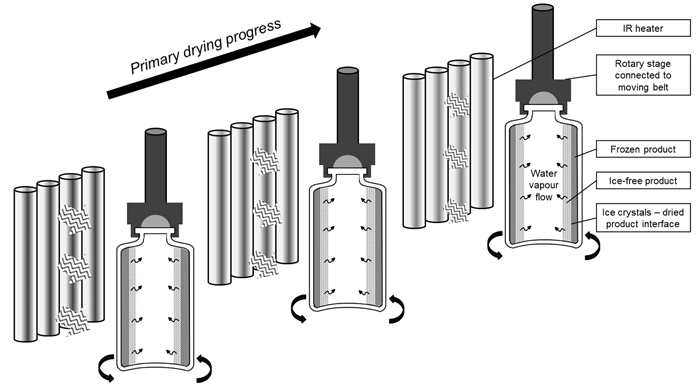
Figure 2: Illustration of the IR assisted continuous primary drying of spin frozen vials, rotating along their longitudinal axis in front of individual IR heaters10
In a conventional freeze-dryer, the sublimated ice and desorbed water is collected using cryogenic ice condensers. For this continuous freeze-drying concept, a condenser system is used allowing it to continuously remove the condensed water. Figure 3 illustrates how an industrial-scale continuous freeze-dryer might look. An extra advantage of this continuous freeze-drying set up is the possibility of implementing process analysers allowing measurement and control at the level of the individual vial. This integrated approach leads to strongly reduced variability of critical quality attributes.
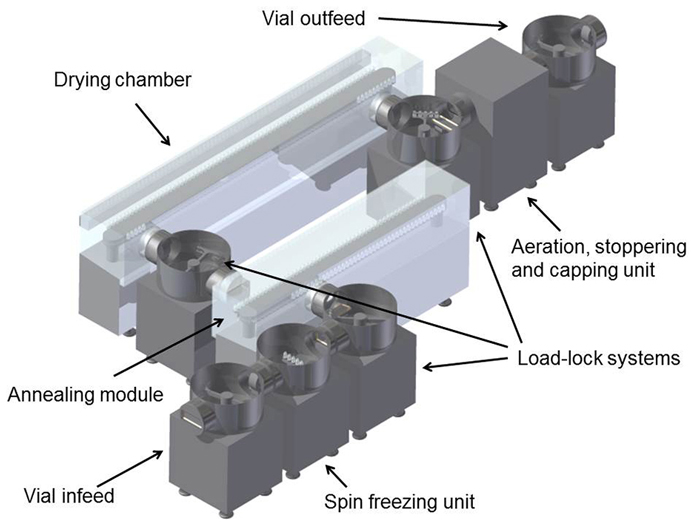
Figure 3: Design drawing of an industrial-scale continuous freeze-dryer
for unit doses
Figure 4 shows schematically that increasing the vial throughput (ie, scale-up) can be simply done by adding parallel lines in the continuous freeze-drying technology modules (ie, Lego principle).
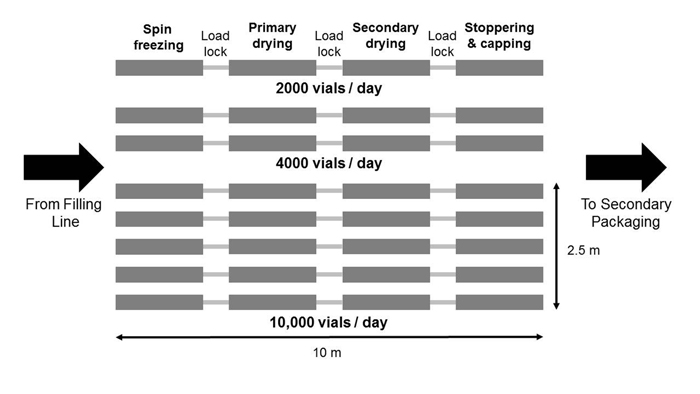
Figure 5: Parallel lines in the continuous freeze-drying technology avoiding scale-up re-optimisation and validation
RheaVita and Ghent University have built two different continuous freeze-drying prototypes:
- A single-vial prototype in which the continuous freeze-drying process of a single vial can be imitated (Figure 5). Identical process conditions can be achieved in this single-vial system as in industrial-scale continuous freeze-dryers, allowing process development at early stages with a very limited amount of drug product material

Figure 5: Single vial prototype
- A GMP-like engineering prototype (Figure 6) where all process modules are integrated and freeze-drying is executed in a continuous fashion. This prototype is engineered around the implementation of needs to create and keep a sterile environment by choosing the proper materials and design principles. Further, the implementation of the relevant PAT tools such as NIR is incorporated and the implementation of the results from mechanistic modelling leads to optimal process conditions.
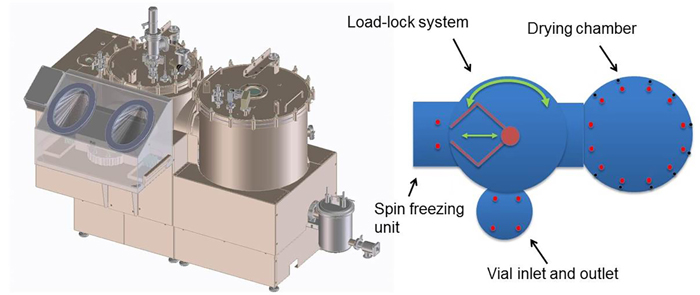
Figure 6: GMP-like engineering prototype
References
- Burns, L. The biopharmaceutical sector’s impact on the US economy: analysis at the national, state and local levels. 2009. www.archstoneconsulting.com/biopharmapdf/report.pdf
- Gieseler H. Insights in lyophilization 2012. Current best practices & research trends. 2012. Antwerp.
- Pikal MJ. Freeze Drying. Encyclopedia of pharmaceutical technology. 2002;1299-1326.
- Kasper J, Friess W. The freezing step in lyophilization: physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. European Journal of Pharmaceutics & Biopharmaceutics. 2017;78:248-263.
- Van Bockstal PJ, Mortier STFC, Corver J, Vervaet C, Nopens I, Gernaey KV, De Beer T. Quantitative risk assessment via uncertainty analysis in combination with error propagation for the determination of the dynamic Design Space of the primary drying step during freeze-drying. European Journal of Pharmaceutics & Biopharmaceutics. 2017;121:32-41.
- International Conference on Harmonization (ICH) of technical requirements for registration of pharmaceuticals for human use, Topic Q8(R2): Pharmaceutical Development. 2009. Geneva.
- United States Food and Drug Administration (FDA), Guidance for industry PAT – A framework for innovative pharmaceutical manufacturing and quality assurance. 2004.
- United States Food and Drug Administration (FDA), Pharmaceutical CGMPs for the 21st century – a risk based approach. 2004.
- Van Bockstal PJ, De Meyer L, Corver J, Vervaet C, De Beer T. Noncontact infrared-mediated heat transfer during continuous freeze-drying of unit doses. Journal of Pharmaceutical Sciences. 2017;106(1):71-82.
- Van Bockstal PJ, Mortier STFC, De Meyer L, Corver J, Vervaet C, Nopens I, De Beer T. Mechanistic modelling of infrared mediated energy transfer during the primary drying step of a continuous freeze-drying process. European Journal of Pharmaceutics & Biopharmaceutics. 2017;114:11-21.
Biography
 PIETER-JAN VAN BOCKSTAL obtained his MS in drug development (2013) from Ghent University, Belgium. Currently, he is pursuing his PhD at the same university under the supervision of Prof Thomas De Beer. His research project aims at developing a continuous and controlled freezedrying technology for unit doses based on noncontact energy transfer via infrared radiation.
PIETER-JAN VAN BOCKSTAL obtained his MS in drug development (2013) from Ghent University, Belgium. Currently, he is pursuing his PhD at the same university under the supervision of Prof Thomas De Beer. His research project aims at developing a continuous and controlled freezedrying technology for unit doses based on noncontact energy transfer via infrared radiation.
 JOS CORVER holds a degree in applied physics in the field of rheology and physical transport phenomena. After a rich career in various industries, he started his own company in 2011, RheaVita, which strives for innovative improvements in pharmaceutical processing, specifically in the field of continuous freeze-drying.
JOS CORVER holds a degree in applied physics in the field of rheology and physical transport phenomena. After a rich career in various industries, he started his own company in 2011, RheaVita, which strives for innovative improvements in pharmaceutical processing, specifically in the field of continuous freeze-drying.

We are looking at a freeze dryer having temperature range of -60 to +65 deg C , shelf size 550 mm *450 mm, distance between shelf 100 mm, 8 no of shelf, condenser temp -80 deg C with 25 lit condenser capacity