Obstacles to success for biosimilars in the US market
Posted: 4 January 2018 | Dr Sarfaraz K Niazi - Karyo Biologics | No comments yet
Since 2007, the EMA has approved 31 biosimilar products1 and refused or withdrawn around five. The FDA, however, since the enactment of the Biologics Price Competition and Innovation Act (BPCIA) in 2009,2 has licensed six products under PHS 351(k) of the Public Health Service (PHS) Act; approved one product under 505 (b)(2) of the Federal Food, Drug, and Cosmetic (FD&C) Act; and sent out four Complete Response Letters (CRLs) to 351(k) Biologics License Applications (BLAs),3 including two applications each for pegfilgrastim and erythropoietin alfa (Table 1).

This commentary is based on my hands-on experience of developing biosimilars in the US and taking them to the 351(k) filing. More specifically, I will share a novel model for developing biosimilars that appears promising in overcoming many obstacles identified herein.
Obstacle 1: understanding the guidance
FDA writes the guidance to comply with two distinct requirements: statutory and scientific. Statutory requirements include analytical similarity, PK/PD, immunogenicity and animal toxicology, which the BPCIA mandates must be met based on scientific requirements that remain flexible and negotiable. I suggest that developers read the BPCIA carefully before reading the guide. One might be surprised to note that the FDA does not mention ‘phase 1’ or ‘phase 3’ in its guidance because human studies for biosimilars are intended to provide a comparative profile, not a measure of efficacy. This distinction is important, yet difficult to embed in the minds of scientists who are used to traditional phase 1-3 studies. Concessions such as using a development lot for a phase 1 study are not available for PK/PD studies for biosimilars, as these must be at-scale GMP lots.
The FDA uses the terms, ‘no residual uncertainty’ and ‘step-by-step’ approach, carefully and in a structured format. If you have resources that allow you to jump the steps, you should find that you are awarded a CRL; never make the mistake of doing what should not be done, such as unnecessary studies in patients.
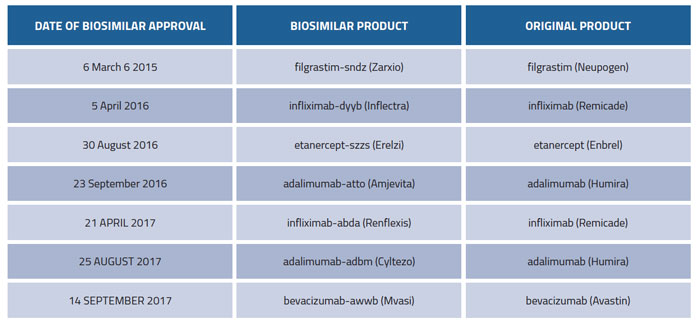
Table 1: FDA approved biosimilars
Obstacle 2: not knowing your molecule
Biosimilar development requires a deeper understanding of the molecule of choice to replicate all limitations and all ‘imperfections’ of the reference product. With a new molecule, you take whatever you have and prove its safety and efficacy; in the case of biosimilars, you need to replicate, even if a feature is not clinically relevant, using a technology that may be protected by hundreds of patents designed to prevent you producing the same molecule. These scientific constraints are better addressed when the development work is outsourced to qualified teams that may not be possible to gather in-house. I strongly urge developers to explore the power of partnering with CDRMOs.
Obstacle 3: ignoring importance of freedom to operate
As of today, every product approved by the FDA is under litigation.4 An extensive freedom to operate analysis will teach that developers should concentrate on a wide choice of potential candidates with a market of over $120bn that have a lesser risk of litigation. Teaching science to the legal team and law to the scientists does not work well; however, there are several expert teams available to conduct these determinations. I recommend outsourcing this work, rather than doing it internally and getting constrained by the available expertise.
Obstacle 4: reliance on patient studies
The reason that the FDA does not mandate a patient study for the approval of biosimilars has its roots in the statutory declaration in CRF 320.25 (a) that, “no unnecessary human research should be done”. When this statute was established for the bioequivalence testing of chemical drugs, the prospect of highly potent biosimilar antibodies was not envisioned; today, the declaration applies more to biosimilars as we develop highly potent and immunogenic products whose testing in humans is riskier. In addition, we now face a new conundrum5 where it has become more difficult to find patient populations on whom to test drugs, causing delays and escalating costs in the development of biosimilars. Developers should aim to avoid all studies in patients, and that requires a different approach to designing other similarity testing studies. A fingerprint-like molecule is least likely to invoke any need for studies in patients.
Obstacle 5: underestimating interchangeability plan
The FDA allows two types of approvals for biosimilars and one of these grants permission to interchange without consultation with the prescriber – an interchangeability designation that requires, by statute, a patient study to demonstrate switching and alternating, demonstrating no less efficacy and no more safety issues. Given the dearth of patients, particularly for anticancer drugs, developers face difficulties in managing patient studies and, as a result, I see fewer applications for the interchangeable status coming to the FDA. My advice: stay away from interchangeable filings if you want a fast-to-market entry. My Citizens Petition to the FDA questions the scientific rationale of approval once the FDA determines that there is ‘no clinically meaningful difference’.
Obstacle 6: not understanding side-by-side testing
Safety and efficacy are tested side by side, with highly complex statistical modelling to prove similarity. How does one determine that there remains no ‘residual uncertainty’? How does one design studies that are sensitive to demonstrating differences rather than similarity? For example, the choice of dosing level in animal toxicology studies, whether for rodents or other mammals, should be within the escalating range of the sigmoid curve. The three statistical tiers in analytical similarity testing are not intended to make one test more important than the other. The tier 1 and tier 2 tests remain fraught with mathematical difficulties that the FDA still needs to fix. A tier 1 test where the reference product has very small standard deviation will always fail, and a tier 2 test will fail if the biosimilar products have fewer degradants or aggregates. The FDA does not allow non-inferiority testing in analytical similarity. Still, the developer needs strong statistical expertise to develop analytical similarity testing protocols. Similarly, clinical studies cannot be overpowered to obviate deficiencies in the design or testing of drugs or antibody levels. The FDA is still improving its guidance on statistical testing; the developer is advised to read the reports that the FDA has made public while approving biosimilar products.
Obstacle 7: GMP compliance
Unlike in the development of a new product, the FDA expects all testing to be done using at-scale lots in a GMP environment and in a CFR Part 11 compliant laboratory. This requirement comes because there is no mandatory patient trial anticipated to establish safety and efficacy of the product. The FDA guidance on GMP for investigational drugs6 is interpreted differently for biosimilars, and the FDA can audit the facility for full GMP compliance in response to an Investigational New Drug (IND) filing – an aspect that many new developers fail to realise. It is for this reason that biosimilar development works better when development work is outsourced, rather than creating a fully integrated traditional facility. Outsourcing also allows developers to bring in creative technologies7 to obtain lower cost of goods sold. In my calculations, it is more efficient and less costly to work with CDRMOs than doing the work internally.
Obstacle 8: FDA meetings
Formal meetings with the FDA are designed to address the issues of ‘residual uncertainty’, rather than function as teaching lessons for developers. A well-informed developer should be able to write meeting requests that result in the definite closure of issues rather than seek general advice. The FDA has made public its evaluation of the first biosimilars approved, and this information should be sufficient to write appropriate protocols, establish analytical similarity testing, select an animal toxicology model, etc. The goal of Type 2 meetings is to come out ready for the Type 3 meeting without going back to another Type 2, as most developers have done recently. Another piece of information for small companies is that while the FDA may waive the filing fee for your first application, the fee for meetings cannot be waived according to the 2017 revision of the Biosimilar User Fee Act (BsUFA). Generally, consultants hired to write and manage FDA applications have little experience and mistake this exercise for a validation of plans – the goal is to negotiate terms, not just shake hands.
References
- www.ema.europa.eu
- www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/ucm216146.pdf
- www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/default.htm
- www.healthlawpolicymatters.com/wp-content/uploads/sites/8/2016/08/Status-of-Biosimilar-A pplications-August-2016.pdf
- www.nytimes.com/2017/08/12/health/cancer-drug-trials-encounter-aproblem- too-few-patients.html?_r=0
- www.fda.gov/downloads/drugs/guidances/ucm070273.pdf
- www.niazi.com/bioprocessing
Biography
SARFARAZ K NIAZI, PhD, SI, FRSB, FPAMS, FACB, is the Founding Executive Chairman and CSO of Karyo Biologics, LLC, the Founder of Adello Biologics, LLC, a fully integrated US biosimilar company, and Adjunct Professor of Biopharmaceutical Sciences, University of Illinois, Chicago. He has over two decades of hands-on experience in taking biosimilars to market globally. He has authored dozens of major books about biosimilars, bioprocessing, pharmaceutical formulations and regulatory management.
Issue
Related topics
Related organisations
European Medicines Agency (EMA), U.S. Food and Drug Administration (FDA)