

Accounting for risk when designing chemical safety assessments for pharmaceutical packaging systems
Posted: 22 March 2018 | Dennis Jenke - Triad Scientific Solutions | 1 comment
Until drug products can be directly manufactured at their point of use, they will be packaged in container closure systems for storage between manufacturing and use. Until packaging systems for drug products are chemically inert, interactions between a drug product and its packaging will remain the rule and not the exception.

A drug product is formulated to provide the maximum therapeutic benefit. When the drug product interacts with its packaging, it is chemically altered, potentially diminishing its therapeutic benefit. For example, chemical substances may leave the packaging system and accumulate in the drug product as foreign impurities (leachables).
Intuitively, the presence of leachables in a drug product is sub-optimal, as it is best that the drug product be as pure as possible. On a practical level, leachables may adversely affect the product’s therapeutic benefit. For example, mutagenic leachables may represent a patient health hazard.
It is a global regulatory requirement that a packaging system’s potential adverse effect on patient safety be addressed. This necessary chemical assessment may include testing of either the packaging system for extractables (E) as potential leachables and/or the drug products for leachables (L). However, due to the great diversity in drug products and packaging systems, the rigour and amount of chemical assessment required to qualify a packaged drug product varies across drug products and packaging systems. To establish the proper type and amount of testing, a risk-based approach can be adopted, where the rigour and amount of E&L testing depends on the risk that: (a) leaching could occur (likelihood of an effect); (b) if leaching did occur, that leachables would adversely affect drug product suitability (severity of the effect). The lesser the risk, the lesser the amount and rigour of chemical testing required. The greater the risk, the more extensive and rigorous the required chemical testing.
Establishing risk
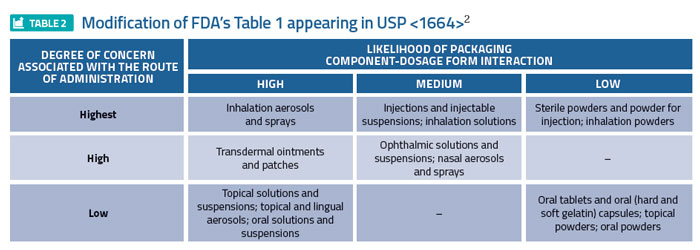
Global regulators have established means for defining the risk associated with a drug product/packaging system interaction, specifically addressing leaching. For example, the FDA’s 1999 Container Closure Guidance1 assigns various drug product dosage forms a relative level of risk, based on the likelihood that a packaging/dosage form interaction could occur, and the degree of concern associated with the route of administration (Table 1). For example, inhalation aerosols and solutions are established as the highest risk dosage forms while oral tablets are established as the least risky dosage form. A somewhat modified form of Table 1 appeared in USP <1664>2 (Table 2), reflecting a risk downgrade for certain dosage forms based on a more recent evaluation of risk factors.
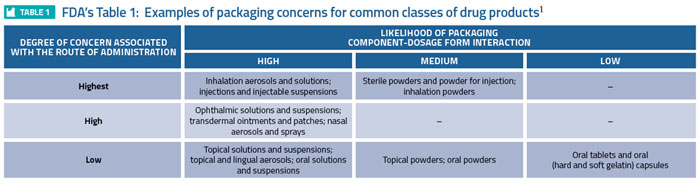 Consistent with the risk defined in Table 1, Table 2 of the FDA guidance provides the means of establishing the level of chemical (and biological) assessment required, as it correlates dosage forms with various testing strategies. For example, extensive testing, including USP Biological Reactivity Testing and possibly extraction/ toxicological evaluation, is necessary to establish that packaging used for injections and injectable suspensions has an acceptably small adverse effect on patient health (known as Case 2s).1 Similarly, the Immediate Packaging Guidelines of the European Medicines Agency (EMA)3 establish the means by which various dosage form packaging systems must be chemically assessed. While the considerations that went into the guidelines and the outcome of applying them are likely similar to those of the FDA Container Closure Guidance, the guidelines establish and manage risk via the use of a flow chart (versus the use of tables and text). In either case – guidance or guidelines – risk assessment drives the rigour and magnitude of chemical testing required to establish that a packaged drug product is safe, at least from a leachables perspective.
Consistent with the risk defined in Table 1, Table 2 of the FDA guidance provides the means of establishing the level of chemical (and biological) assessment required, as it correlates dosage forms with various testing strategies. For example, extensive testing, including USP Biological Reactivity Testing and possibly extraction/ toxicological evaluation, is necessary to establish that packaging used for injections and injectable suspensions has an acceptably small adverse effect on patient health (known as Case 2s).1 Similarly, the Immediate Packaging Guidelines of the European Medicines Agency (EMA)3 establish the means by which various dosage form packaging systems must be chemically assessed. While the considerations that went into the guidelines and the outcome of applying them are likely similar to those of the FDA Container Closure Guidance, the guidelines establish and manage risk via the use of a flow chart (versus the use of tables and text). In either case – guidance or guidelines – risk assessment drives the rigour and magnitude of chemical testing required to establish that a packaged drug product is safe, at least from a leachables perspective.
Missing dimensions of risk?
In 2006, the Product Quality Research Institute (PQRI) published its Best Practice Recommendations for Extractables and Leachables in Orally Inhaled and Nasal Drug Products.4 A key contribution of this text was the concept of the Safety Concern Threshold (SCT), defined as “the threshold below which a leachable would have a dose so low as to present negligible safety concerns from carcinogenic and noncarcinogenic toxic effects”.
Considering leachables, a patient’s exposure to a leachable (dose, Dl) is established as the product of the concentration of the leachable in the drug product (Cl) and the drug product’s daily dose volume (Vd): Dl = Cl x Vd
If there is a dose of a leachable that establishes what is essentially a safety threshold (the SCT), then it is clear that the safe concentration of a leachable is
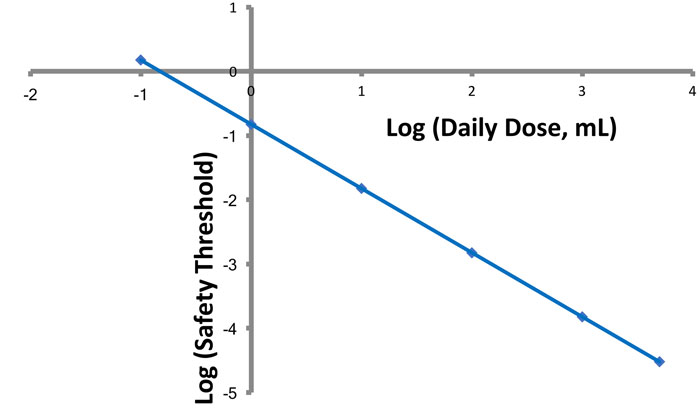
Figure 1: Effect of daily dose volume on an analytical threshold. The numerical value for an analytical threshold decreases in direct proportion to the increase in daily dose volume.
inversely proportional to the daily dose volume (Figure 1). The smaller the value of Vd, the larger the value of the leachable’s safe concentration.
It is well known that drug product dosage forms vary greatly in terms of their Vd value (Figure 2). Thus, Vd is an appropriate factor to consider in establishing the safety risk associated with various dosage forms. Furthermore, this factor may be undervalued in the previously cited FDA and EMA risk evaluation processes.
Consider, for example, the broad category of drug products termed non-solid dosage forms for parenteral administration defined in the EMA guideline and injections in the FDA guidance. This broad product class includes drug products (for example, vaccines) with a very low daily dose volume (typically measured in fractions of a millilitre) and drug products (for example, large-volume parenterals for nutrition therapy) with a very large daily dose volume (typically measured in multiple litres).
It is clear that these dosage forms, although belonging to the same dosage form family, have greatly differing risks related to the safety impact of leachables, as the widely different patient exposure to leachables is dictated by their widely different daily dose volumes. It can be argued, then, that in a risk-based strategy, the testing of the vaccine and its packaging system would be less extensive and rigorous than the testing of the LVP and its packaging system.
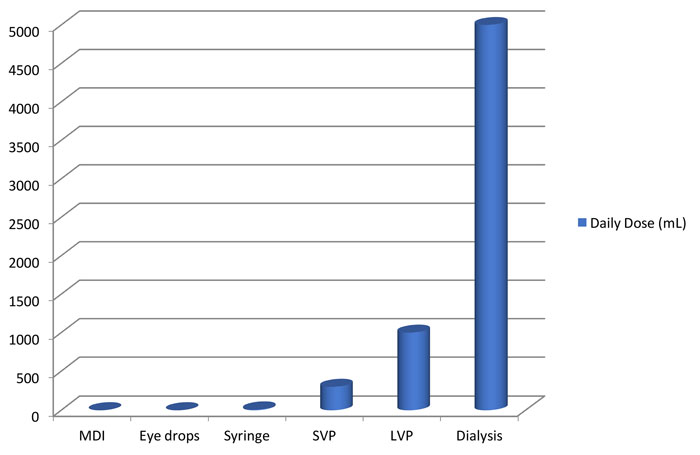
Figure 2: Daily dose volumes for general classes of pharmaceutical drug products. Daily dose volumes can vary from less than 1mL to tens of litres.
Another factor to consider in assessing risk for the purpose of designing efficient chemical assessments is the drug product’s duration of clinical therapy. Simply stated, and as noted in the M7 ICH Guideline for the assessment and control of mutagenic impurities in pharmaceuticals,5 the “cancer risk of a continuous low dose over a lifetime would be equivalent to the cancer risk associated with an identical cumulative exposure averaged over a shorter duration”. Thus, for example, the safety risk associated with leachables would be lower for a flu vaccine dosed once a year versus a dialysis solution that is used daily over the patient’s lifetime (considering only duration and frequency of exposure), suggesting that a less rigorous chemical assessment of the vaccine and its packaging is appropriate.
Take away message
The regulatory guidance documents and guidelines are a useful and necessary part of a risk-based strategy for chemical assessment, including both extractables testing of packaging systems and leachables testing of drug products. However, use of the risk assessment tools in these documents does not necessarily constitute the complete risk assessment process; rather, these tools represent the first step in the process and are logically followed by a consideration of additional relevant and applicable dimensions of risk. So doing, leverages risk dimensions that are shared by all drug products within a dosage form classification but recognises the considerable differences between those drug products, thereby resulting in effective and efficient chemical assessments.
Wondering out loud
I sometimes wonder, as we develop chemical characterisation strategies for packaging, whether we are developing strategies to meet yesterday’s challenges or today’s realities. It is possible that in the past there may have been a poorer awareness of the potential adverse effect of packaging on drug product use suitability (including patient safety), leading to the litany of issues that have arisen and which have been documented and addressed. However, with each issue resolved and with heightened vigilance with respect to potential safety issues being exercised during packaging and drug product development, it becomes ever more likely that we have reached a state approaching the ideal where quality is built, rather than tested, into the product.
Drug and packaging development scientists have partnered with suppliers of packaging materials to address and resolve issues, both known and anticipated. The resolution of three of the more well-known E&L issues attests to that. The use of Teflon-coated stoppers to resolve a patient safety issue for the EPREX drug product (which arose when a formulation change increased the leaching of a reactive curing agent)6 is one such example. The modification of the forming process for glass syringes, directed towards minimising tungsten residuals associated with the pin that is used to form the pin’s cavity, was undertaken to reduce unfortunate outcomes related to protein-containing drug products.7 Lastly, reducing (or eliminating) a specific antioxidant from plastic materials used in multi-layered bioreactor bags, was used to address an antioxidant-related leachable that reduced cell culture yields from certain sensitive cell lines.8
These are but a few examples of the industry’s ongoing march towards the goal of reducing product suitability use issues related to leachables by intelligent product design, or, when necessary and appropriate, re-design.
While it is far too soon to declare that we have solved all the issues associated with leachables and can therefore do away with certain aspects of chemical safety assessment, now may be the right time to leverage the progress made to ensure that the right studies are performed to produce the right results with the right levels of time and effort.
References
- Guidance for Industry: Container Closure Systems for Packaging Human Drugs and Biologics-Chemistry, Manufacturing, and Controls Documentation. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics and Research (CBER). May 1999.
- USP <1664>, Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery System. United States Pharmacopeia; First Supplement to USP 38-NF 33, pp. 7181-7193, official from August 1, 2015.
- European Medicines Agency; Committee For Medicinal Products for Human Use (CHMP), Committee for Medicinal Products for Veterinary Use (CVMP). Guideline on Plastic Immediate Packaging Materials. CPMP/QWP/4359/03. London; 19 May 2005.
- Safety Thresholds and Best Practices for Extractables and Leachables in Orally Inhaled and Nasal Drug Products, PQRI Leachables and Extractables Working Group. Product Quality Research Institute. 2006.
- M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. Guidance for Industry. U.S Department of Health and Human Services, Food and Drug Administration. May, 2015.
- Pang J, Brown J, Labrenz S, Villalobos A, Depaolis AS, Gunturi S, Grossman S, Lisi P, Heavner GA. Recognition and identification of UV-absorbing leachables in EPREX® Pre-filled syringes: An unexpected occurrence at a formulation-component interface. PDA J Pharm Sci Technol. 2007;61(6): 423-432.
- Liu W, Swift R, Torraca G, Nashed-Samuel Y, Wen Z-Q, Jiang Y, Vance A, Mire-Sluis A, Freund E, Davis J, Narhi L. Root cause analysis of tungsten-induced protein aggregation in pre-filled syringes. PDA J Sci Technol. 2010;66(1):11-19.
- Hammond M, Nunn H, Rogers G, Lee H, Marghitoiu AL, Perez L, Nashed-Samuel Y, Anderson C, Vandiver M, Kline S. Identification of a leachable compound detrimental to cell growth in single-use bioprocess containers. PDA J Sci Technol. 2013;67(2):123-134.
 DR DENNIS JENKE is Chief Executive Scientist at Triad Scientific Solutions. He is the author of Compatibility of Pharmaceutical Solutions and Contact Materials, Safety Considerations Associated with Extractables and Leachables, and a contributing author to the Leachables and Extractables Handbook. Dr Jenke is a member of numerous industry groups whose charter is to establish best demonstrated practices in the area of material/solution compatibility. Dr Jenke can be contacted at dennisjenke@ triadscientificsolutions.com.
DR DENNIS JENKE is Chief Executive Scientist at Triad Scientific Solutions. He is the author of Compatibility of Pharmaceutical Solutions and Contact Materials, Safety Considerations Associated with Extractables and Leachables, and a contributing author to the Leachables and Extractables Handbook. Dr Jenke is a member of numerous industry groups whose charter is to establish best demonstrated practices in the area of material/solution compatibility. Dr Jenke can be contacted at dennisjenke@ triadscientificsolutions.com.





Very good overview of the risk based approach and consequences for E&L testing. One thing that tends to be forgotten is the big question: ‘Did (or could) anything change since I did the assessment ?’ Unfortunately the answer is ‘very likely’. There is a very big assumption, that ingredients (polymers and colors) for plastic packaging materials do not change. This is false. As Dennis points out, risk is managed by thorough assessment, so it is very surprising when talking to packaging development engineers , that there is generally poor understanding of what goes into plastic materials the risk of change. Unfortunately this is too often driven by, the view that the ingredient(s) have ‘food contact’ declarations, and I have a change control agreement and ‘DMF’ from my packaging supplier – so it is OK. Behind the direct packaging supplier, there is a different world, which probably the regulator understands better than the industry in general. This is why some aspects of the new USP could generate some interesting discussions.