Pharmaceutical QbD: Omnipresence in the product development lifecycle
Posted: 18 April 2017 | Mahfoozur Rahman (Sam Higginbottom Institute of Agriculture), Sagar Suman Panda (Roland Institute of Pharmaceutical Sciences), Sawar Beg (Jubilant Generics Limited) | No comments yet
The concept of Quality by Design (QbD) was implemented by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and United States Food and Drug Administration (FDA) around a decade ago, with the aim of adopting systematic approaches for developing high-quality pharmaceutical products for patients’ benefit and to meet regulatory needs…

QbD does not just facilitate product and process understanding, it also increases regulatory flexibility, enabling faster approval of products with reduced numbers of recalls and rejects. With the ascent of time, a series of guidances have been published by ICH and endorsed by the FDA for harmonising the holistic implementation of QbD paradigms in pharmaceutical manufacturing. This has certainly revolutionised traditional testing-intensive pharmaceutical manufacturing.
QbD has brought enormous benefits to the development of drug substances, analytical methods, dissolution testing, stability studies, bioequivalence testing and clinical trials. Regulatory agencies are encouraging the pharmaceutical industry to holistically implement QbD in all of these stages to reduce product development times, and accelerate regulatory review and approval processes. This article endeavours to provide an overview on vital precepts of QbD paradigms during different stages of development for efficient pharmaceutical manufacturing to produce a robust product with the desired safety and efficacy.
Introduction
For decades the pharmaceutical industry has been continuously engaged in developing new drug products and generics to cater for healthcare demands and serve the patient community. However, owing to the involvement of active drug substance(s), multiple functional/nonfunctional excipients and manufacturing processes that lack automation, pharmaceutical products are facing a high degree of variability in their quality, which has become a major topic of debate.
In 2002 a news article published in The Wall Street Journal stated:
“although the pharmaceutical industry has little secret…, yet its manufacturing standards lag far behind potato chips and laundry soap makers”,
while another article published in 2007 mentioned that the:
“pharmaceutical industry is producing six-sigma products using three-sigma processes”1,2.
These news articles shook the entire pharma world, but demonstrate that assuring consistency in the quality of drug products is a major point of concern, and something that is taken seriously by the federal regulatory agencies.
To revolutionise quality standards, regulatory agencies started focusing on the pharmaceutical manufacturing standards and approval process. In 2003 the FDA proposed the first major quality initiative in the form of “Pharmaceutical cGMPS for the 21st Century-A Risk-Based Approach” to safeguard patients’ health with the promise of delivering high-quality drug products3. This demonstrates that systematic science- and risk-based approaches were starting to be adopted during product development practices. Later, the International Conference on Harmonization (ICH) instituted a series of guidances including Q8 (Pharmaceutical development), Q9 (Quality Risk Management) and Q10 (Pharmaceutical Quality System) for harmonising the adoption of quality principles by industry and regulatory agencies4-6. All three guidances are interrelated, although Q8 primarily focuses on precepts and fundamental elements of QbD.
The concept of quality is not new to the world and has been important in manufacturing practice since the 1980s. Motorola was the first company to implement the use of systematic quality principles into manufacturing practice, which later percolated into several technology-driven industries including information technology, telecommunication, automobile and aeronautic7. However, such principles were adopted in pharmaceutical development much later. After the institution of ICH Q8-Q10 guidances, the FDA started coercing the pharma industries to adopt QbD paradigms to improve their manufacturing standards.

Figure 1: Quality triology as per the Juran’s quality philosophy.
Based on the principles of six-sigma, the QbD concept was first outlined by an American quality expert, Joseph M. Juran, through his publications and book, “Juran on Quality by Design”8. He described the concept in the form of Juran’s triology, where quality is considered as a dynamic trait and undergoes continuous improvement by the act of conscious intent with apt planning and control. Figure 1 illustrates the concept of Juran’s quality triology, which aims to deliver products with the goal of quality by planning but not by chance9. Moreover, Juran also enunciated the philosophy of building quality into the products by design to avoid the traditional practice of quality by testing.
- Quality Planning
This involves planning the quality at the very first stage based on the “voice of customer”
- Quality Control
The variability in product and process attributes needs to be controlled by adopting risk-based statistical tools for monitoring the end-product quality
- Quality Improvement
This is a continuous process, which is achieved throughout the product life cycle by regulating the controls
Pharmaceutical QbD: Fundamental principles
Based on Juran’s concept, pharmaceutical QbD primarily takes the approach of envisioning and planning product quality based on predefined objectives. It increases product quality and robustness; increases customer satisfaction; reduces defects, recalls and rejects, thus ultimately providing significant savings of resources such as time, effort and cost. Beyond these, there are several long-term benefits of QbD10,11. Below provides a detailed account on the benefits of QbD:
- Reduces development time
- Minimises product cost by multiple times
- Helps in effective troubleshooting
- Facilitates timely launch of the products
- Increases consumer generic acceptance
- Monitors regulatory oversight with greater flexibility
- Maintains product life cycle with greater ease
The implementation of QbD in an industrial scenario can be achieved through five key steps, which are discussed in the following sections12,13. These steps work alongside the principles of six sigma, in order to develop high-quality robust drug products with enhanced product and process understanding. Figure 2 portrays the key five steps of QbD on which the product development process is based. These steps are highly important and mandatory in the form of minimal requirements for regulatory approval of drug products.
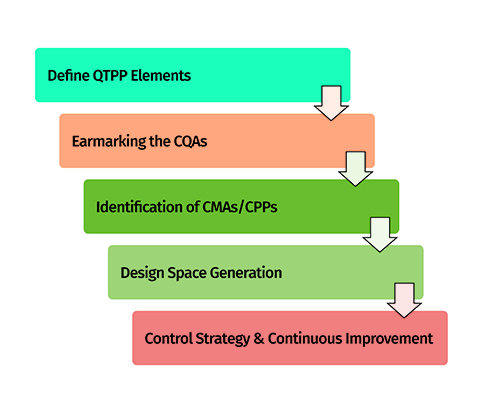
Figure 2: Five-steps approach for implementation of QbD.
Step 1: Defining the objectives and quality planning
Systematic planning among product development team members is essential for identifying product development objectives before development begins. As per the QbD approach, it is referred to as a quality target product profile (QTPP), which summarises the quality characteristics of the target product. QTPP primarily includes vital product features such as dosage form type, dose, route of administration, pharmacokinetics and stability, which need to be strictly monitored throughout the product development exercise14.
Step 2: Identification of product quality attributes
After defining QTPP, the key quality attributes directly associated with product quality are considered while identifying the key vital critical quality attributes (CQAs). The CQAs are also referred to as potential indictors of the product quality performance and directly related to QTPP11,13. These are identified after concerted team efforts to suitably justify their need with respect to their importance in benefitting patients. Instances of CQAs include physical, chemical, biological and microbiological attributes of the drug product, which need to be critically monitored with respect to their defined specifications to avoid inconsistency in the product quality4. As per the QbD-based approach, the desired goals for CQAs are achieved using DoE and QRM principles to identify the critical material attributes (CMAs) or critical process parameters (CPPs), followed by optimisation of them using multivariate experimental designs (Figure 3).
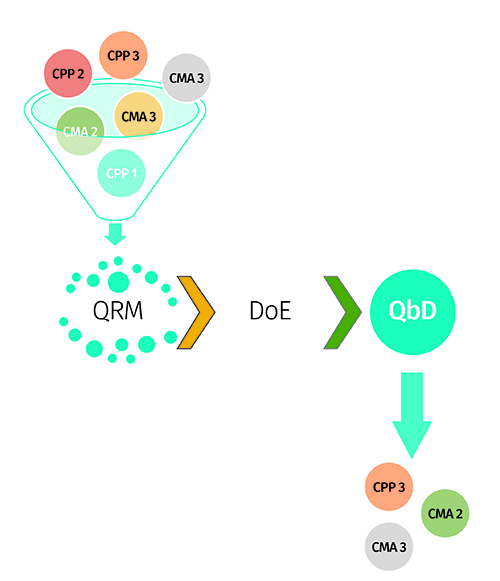
Figure 3: Amalgamation of QRM and DoE approach for factor screening and optimisation.
Step 3: Establishment of specifications on target quality
The approach of establishing set specifications for the target product’s quality is carried out by identifying a suitable design space. It is a multidimensional region generated between the CMAs or CPPs, which have direct influence on the product CQAs. Risk assessment or factor screening studies are primarily used for identifying the “vital few” CMAs/CPPs from the plausible “so many”10. Figure 4 depicts the critical steps involved during risk assessment and risk mitigation exercises in an industrial set-up. Risk estimation matrix and failure mode effect analysis techniques are primarily used for risk assessment, while screening experimental designs, such as Taguchi, Plackett-Burman and fractional factorial designs, are used for factor screening purposes12.
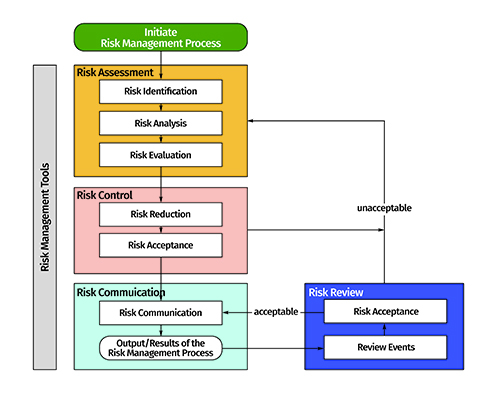
Figure 4: Risk identification, risk assessment and risk mitigation as per the QRM approach (adapted from ICH Q9 guidance: www.ich.org).
Figure 5 shows a pictorial flow chart depicting the importance of the factor screening exercise during QbD-based product development and optimisation. Moreover, response surface designs such as full factorial, central composite, Box-Behnken, optimal and mixture designs are used for optimisation of the CMAs/CPPs.
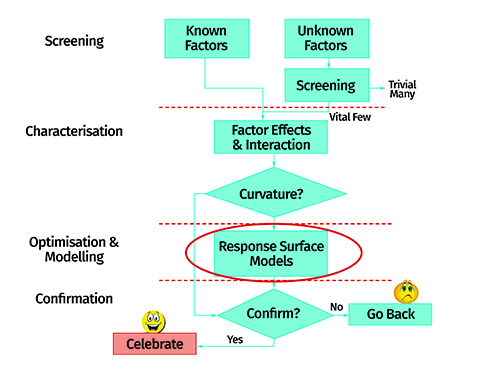
Figure 6: Schematic flow layout of the response surface optimisation method.
Figure 6 portrays the steps encountered during response surface optimisation-based product development. The major goal of such optimisation is to identify the design space. The design space is demarcated within the defined specification limits for the CQAs.
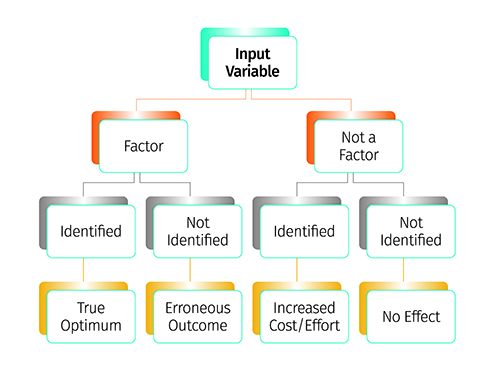
Figure 5: Factor screening study depicting the identification of influential factors.
Following submission of the product development report, the design space is approved by the regulatory agencies – thus it is also termed a “proven acceptable range (PAR)”. Movement within the approved specifications of the design space is allowed without any prior notification(s) to the regulatory agencies, while movement outside the design space is considered as “change” and requires further regulatory approval.
Regulatory agencies are encouraging the industry to identify a control space (also called “normal operating range”) within the design space, on the basis of in-house specifications, to work in a narrower and more constrained region13. Figure 7 pictorially demonstrates the layout of different spaces and their regulatory importance during product development and approval processes. Various types of design space tend to exist during product development cycles, which include lab scale, pilot scale and commercial scale design spaces. Among these, only commercial scale design space has real-time importance for regulatory review and approval of the products14.
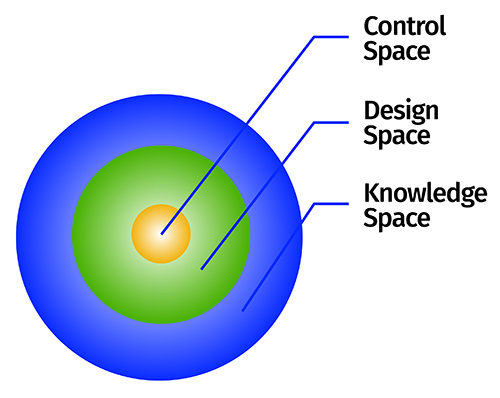
Figure 7: Depiction of various spaces as per the QbD concept.
Step 4: Validation of quality specifications
Evaluation of the robustness of design space is carried out by performing ‘edge of failure’ studies. Since design space established only at commercial scale has regulatory importance for product approval, validation of lab scale design space at pilot, exhibit and commercial scale is considered to be essential. As per the federal demands, the sponsor should demonstrate the robustness of design space at commercial scale followed by product and process understanding14.
Step 5: Quality control strategy and continuous improvement
Based on the entire product development exercise, the firm must establish a control strategy for enabling continuous improvement. These include different types of controls used to ensure the product’s quality, including in-process and end-product testing13. It helps with monitoring the product performance at key stages of a product’s life cycle, including development, technology transfer and commercial manufacturing. Figure 8 illustrates a control strategy during these stages of a product development lifecycle. Postulation of control strategy at these vital stages tends to facilitate change management and reduces its impact on the product’s quality to avoid a quality crisis.
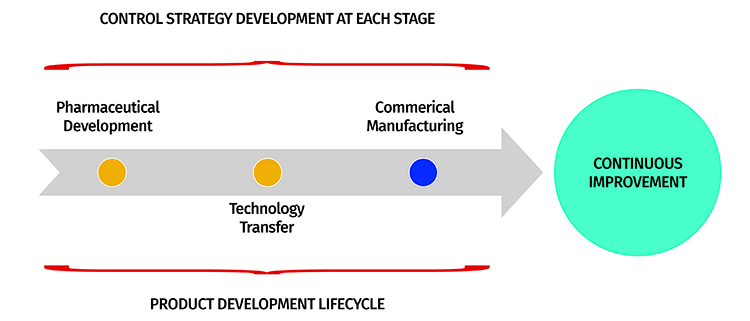
Figure 8: Control strategy flow layout at different product development stages.
Pharma QbD: A lifecycle approach
Besides formulation development, the QbD concept has now permeated into other vital areas of pharmaceutical development including drug substance development, analytical method development, dissolution testing, stability testing, bioequivalence testing, clinical trials, etc.15. It is therefore considered to be omnipresent during each of these vital stages of product development. A formulation scientist can even continue to derive QbD benefits after a product’s commercial launch and post-marketing surveillance. A brief account on QbD implementation at various product development stages is given as follows (also pictorially depicted in Figure 9.
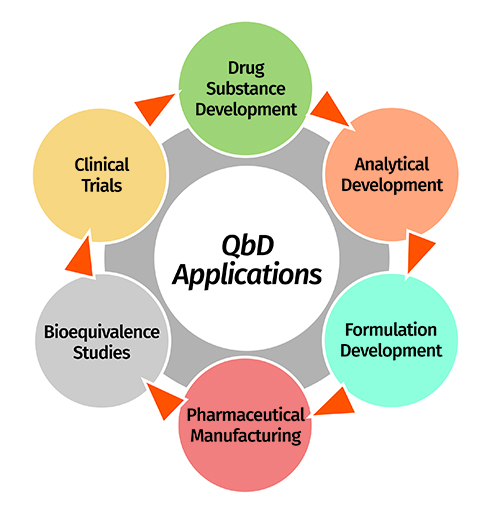
Figure 9: Potential applications of QbD approach in diverse stages of product development lifecycle.
Drug substance and excipient development
The quality of finished products depends on the use of high quality raw materials, which includes drug substance and excipients16,17. Variability in these raw materials produces end-products with highly variable quality, and so implementing QbD principles helps to improve the quality of raw materials supplied by the vendors to pharmaceutical manufacturers. The key CMAs of drug substance include particle size, physical form, polymorphism, moisture content and flow properties, which impact the disintegration, dissolution, compaction and compression characteristics as the CQAs. Likewise, the key CMAs of excipients include particle size, shape, viscosity grade, moisture content, flowability, etc., which also significantly influence the aforementioned CQAs. Moreover, the specifications enlisted in ICH Q11 guidance are applicable to QbD requirements for drug substance development along with critical measures on chemistry, manufacturing and control processes18.
Analytical method development
Use of a robust analytical method is essential for hassle-free monitoring of quality and accelerating the product development process. QbD principles, in this regard, facilitate the development of highly robust analytical methods based on predefined objectives or quality target method profile. Critical method variables (CMVs) are identified on the basis of critical analytical attributes for attaining enhanced method performance for continuous improvement within the design space19. In a nutshell, QbD provides a framework to identify the source of variability and reduce it during analytical method development, thus going beyond the traditional ICH recommended validation process20. Although analytical QbD is not mandatory and no regulatory guidelines yet exist, the federal agencies are encouraging the pharmaceutical industry to adopt the implementation of systematic QbD paradigms for improving analytical understanding.
Dissolution testing
Considered as a quality control tool, dissolution testing helps to monitor the drug release profile of dosage forms. This helps the manufacturer to make decisions in qualifying the batches on the basis of release criteria. In this regard, establishment of a discriminating, robust and predictable dissolution method is imperative. A QbD approach provides greater flexibility during dissolution development by optimising the influence of multiple factors, such as media type, composition, volume, apparatus selection and operating conditions on drug release performance. Regulatory agencies today demand highly robust drug products with an in vitro dissolution performance close to that of the reference listed product, so establishing a predictable in vitro/in vivo correlation for the accomplishing biowaiver21.
Stability testing
Implementing QbD principles in stability testing helps to establish specifications (temperature and humidity conditions, packaging materials, etc.) for stability testing of the products and monitoring controls for identifying the product shelf-life, levels of impurities, degradation products, etc.22. As the concept gains importance in product development, the number of long-term regulatory benefits will rise.
Bioequivalence testing
Bioequivalence tests are used for the final assessment of the in vivo product performance and to evaluate the plausibility of matching between the generic product and reference product24. Vital pharmacokinetic metrics such as the Cmax and AUC ratio between the test and reference products must be within the regulatory acceptance limit of 80%-125% for bioequivalence. In this regard, QbD furnishes formulation development by establishing a link between the input product and process parameters to optimise the in vivo performance. The concept has limited regulatory importance owing to the lack of practical understanding in establishing direct correlation between the in vitro and in vivo product performance parameters.
Clinical trials
The application of QbD principles to clinical trials is one of the most recently investigated scenarios. Such an approach examines the design and objectives of a clinical trial protocol by identifying the ‘critical to quality’ factors and managing the impact of risk on trial quality. Since high expenditures are incurred from clinical trials, federal agencies have been emphasising the current opportunities with QbD risk-based monitoring of clinical studies to help bring about the best possible scientific outcomes for product safety and efficacy.
Conclusion
QbD applications that can address multiple challenges and quality issues are increasingly being used at different stages of pharmaceutical product development. An organisation can reap maximal benefits of QbD by collaborating closely with internal departments to work synergistically for holistic product development practices. Regulatory agencies are also encouraging drugs manufacturers to adopt a greater number of science-based approaches that improve the quality of products and provide benefits during question-based review processes.
Declaration: The views expressed herein are the views and opinions of the authors and do not reflect or represent the views of any of the organisations with which the authors are affiliated.
Biographies
 SARWAR BEG, PhD is a Research Scientist at Jubilant Generics Limited, Noida, India. His major areas of research interest include QbD-driven development and characterisation of diverse drug delivery systems. To date he has authored more than 90 research/review publications in several peer-reviewed journals, 15 book chapters, four books, three Indian patent applications with h-index of 16 and several young scientist awards to his credit. Moreover, he is serving as the Regional Editor of Current Nanomedicine, Editorial Board Member of Bioavailability and Bioequivalence Journal and Nanotech Research Letter, and reviewer of several international journals.
SARWAR BEG, PhD is a Research Scientist at Jubilant Generics Limited, Noida, India. His major areas of research interest include QbD-driven development and characterisation of diverse drug delivery systems. To date he has authored more than 90 research/review publications in several peer-reviewed journals, 15 book chapters, four books, three Indian patent applications with h-index of 16 and several young scientist awards to his credit. Moreover, he is serving as the Regional Editor of Current Nanomedicine, Editorial Board Member of Bioavailability and Bioequivalence Journal and Nanotech Research Letter, and reviewer of several international journals.
 MAHFOOZUR RAHMAN is an Assistant Professor at the Department of Pharmaceutical Sciences, Faculty of Health Sciences, Sam Higginbottom Institute of Agriculture, Technology & Sciences (SHIATS), Allahabad, India. He completed his Masters in Pharmacy in 2010 from Jamia Hamdard, New Delhi, India. His major areas of research interest include development and characterisation of nano-size drug delivery systems for inflammatory disorders. To date he has published over 50 publications in peer reviewed journals with H-index of 12 and eight book chapters with various reputed publishers. He is a member of IPGA and editorial board member of various journals.
MAHFOOZUR RAHMAN is an Assistant Professor at the Department of Pharmaceutical Sciences, Faculty of Health Sciences, Sam Higginbottom Institute of Agriculture, Technology & Sciences (SHIATS), Allahabad, India. He completed his Masters in Pharmacy in 2010 from Jamia Hamdard, New Delhi, India. His major areas of research interest include development and characterisation of nano-size drug delivery systems for inflammatory disorders. To date he has published over 50 publications in peer reviewed journals with H-index of 12 and eight book chapters with various reputed publishers. He is a member of IPGA and editorial board member of various journals.
 MR SAGAR SUMAN PANDA is currently working as an Assistant Professor at Roland Institute of Pharmaceutical Sciences, Berhampur, Odisha, India. Mr Panda has seven years of teaching experience in pharmaceutical analysis and major research interest in QbD-based analytical method development and validation. He has over 35 research publications in various peer-reviewed journals, one book on analytical QbD and several conference presentations to his credit.
MR SAGAR SUMAN PANDA is currently working as an Assistant Professor at Roland Institute of Pharmaceutical Sciences, Berhampur, Odisha, India. Mr Panda has seven years of teaching experience in pharmaceutical analysis and major research interest in QbD-based analytical method development and validation. He has over 35 research publications in various peer-reviewed journals, one book on analytical QbD and several conference presentations to his credit.
References
- New prescription for drug makers: Update the plants. The Wall Street Journal. 2002: CCXLII
- Making improvement work in pharmaceutical manufacturing. ISPE Midwest Extended Education and Vendor Day, Overland Park, KS. 2007
- Pharmaceutical cGMPS for the 21st century — A risk-based approach. 2004
- ICH Harmonised Tripartite Guideline. Q8(R2)-Pharmaceutical Development. 2004
- ICH Harmonised Tripartite Guideline. Q9-Quality Risk Management. 2005
- ICH Harmonised Tripartite Guideline. Q10-Pharmaceutical Quality System. 2008
- Bill S. The inventors of six sigma. 2005
- Juran JM. Juran on Quality by Design: The new steps for planning quality into goods and services. Free Press, New York: USA. 1992
- Juran Trilogy: Quality assurance. 2007 Available on line: https://blogs.mtu.edu/ improvement/2014/02/27/the-juran-trilogy/
- Singh B. QbD in Pharma product development life cycle. Chron Pharmabiz. 2013; 28: 72-79
- Singh B, Beg S, Sharma G, Jain A, Negi P. Holistic application of Quality by Design (QbD) for pharma product development excellence and regulatory compliance. Nirma University J Pharm Sci. 2014; 1 (1): 19-35
- Singh B, Raza K, Beg S. Developing “Optimized” drug products employing “Designed” experiments. Chem Ind Digest. 2013; June: 70-76
- Lionberger RA, Lee SL, Lee L, Raw A, Yu LX. Quality by design: concepts for ANDAs. AAPS J. 2008; 10: 268-76
- Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, Woodcock, J. Understanding pharmaceutical Quality by Design. AAPS J. 2014; 16 (4): 771–783
- Singh B. Quality by Design (QbD) for holistic pharma excellence and regulatory compliance. The Pharma Times. 2014; 46 (8): 26-33
- Drakulich A. An enhanced approach to drug-substance development and manufacture. Pharm Tech. 2012; 36 (2): 1-4
- Arnum PA. Quality by Design in excipients. Pharm Tech. 2013; 37 (2): 1-3
- ICH Harmonised Tripartite Guideline. Q11-Development and Manufacture of Drug Substances. 2012
- Bhutani H, Kurmi M, Singh S, Beg S, Singh B. Quality by Design (QbD) in analytical sciences: An overview. The Pharma Times. 2014; 46 (8): 71-75
- Reid GL, Morgado J, Barnett K, Harrington B, Wang J, Harwood J, Fortin D. Analytical Quality by design (AQbD) in pharmaceutical development. Amr Pharma Rev. 2013, Available online at: www.americanpharmaceuticalreview.com
- Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, Hammett T, Billett L, Behn S, Gibb RC, Abrahamsson B. Clinical relevance of dissolution testing in Quality by Design. AAPS J. 2008; 10 (2): 380–390
- Chatterjee S. QbD principles for packaging design and stability: A regulatory perspective. AICHE Annual Meeting. October 17, 2011. Available online at: http://www.cbinet.com/sites/default/ files/files/Krumenaker_QBD_SC%20AICHE%20Mtg%20101011_V2.pdf (Accessed on 17 November 2016)
- Stier E. FDA recommendations for ANDAs based on QbD: A bioequivalence case study. 44ème Congrès International SFSTP. June 6-7, 2012. Available online at: http://congres.sfstp. myevent.evenium.com/uploads/event_member/85118/20ethanstier.pdf
- Sprenger K, Nickerson D, O’Connell AM, Morrison BW. Quality by Design in clinical trials: A collaborative pilot with FDA. Ther Innov Reg Sci. 2012
- Mulinde J. Clinical Quality by Design: FDA point of view. 51st Annual Meeting: DIA 2015, Washington, D.C. Available online at: http://www.fda.gov/downloads/AboutFDA/Centers Offices/OfficeofMedicalProductsandTobacco/CDER/UCM453948.pdf
Issue
Related topics
Analytical techniques, Drug Development, Excipients, QA/QC, Quality by Design (QbD)