Specifications and method/process capability: two sides of the same coin
Posted: 11 September 2018 | Dr Dave P Elder | No comments yet
Specifications are defined as “a list of tests, references to analytical procedures, and appropriate acceptance criteria, which are numerical limits, ranges, or other criteria for the tests described”.1

Specifications establish predefined criteria of the expected quality of a product – Active Pharmaceutical Ingredient (API) or drug product – at that stage of their development. That is, there is, or should be, an expectation that specifications will evolve as process understanding and knowledge2 increases.
Specifications are therefore a compilation of “critical quality standards [CQSs] linked to overall product performance, which are proposed and endorsed by the applicant and reviewed and approved by the appropriate regulatory or pharmacopoeial authorities”.3 Despite decades of “harmonisation” initiatives, it is by no means certain that acceptance by one regulatory authority, eg. Euoropean Medicines Agency (EMA), will result in acceptance or approval by others, eg. FDA or Pharmaceuticals and Medical Devices Agency (PMDA).
Indeed, applicants often see different opinions from within the same EU regulatory region. Hence, the concept of a “universal specification” for a product is often aspirational in nature, rather than a realistic goal.1
The link between specifications and the underlying critical process parameters (CPPs) and critical quality attributes, which are generated as part of a QbD-type (Quality by Design) submission2,4,5 have never been fully articulated, although all are part of the overall control strategy. Some commentators have expressed concern that ICH Q6A was never revised to more fully explain the inter-relationship.3
The application of ICH Q6A1 during development, particularly early phase development, is one of the key challenges facing applicants and reviewers alike. In general, these guidelines were intended to be applied to marketing approval on new products, except for ICH M7(R1).6 This is sensible as knowledge of CQAs at this early stage of development may be very limited. Significant changes will be expected to both the synthesis / process of the drug substance and to the formulation / process of the drug product as they proceed towards market.3 ICH M3(R2)7 does provide some clarity of the original intent. It states in connection with impurities: “The approaches for qualifying impurities and degradants are outlined in ICH Q3A(R2)8 and Q3B(R2).9 If specific studies are warranted to qualify an impurity or degradant, generally these studies are not warranted before Phase III, unless there are changes that result in a significant new impurity profile e.g., “a new synthetic pathway, a new degradant formed by interactions between the components of the formulation”.
However, the reality is that ICH Q6A guidance is often widely used during clinical development by both industry and regulators alike; often inappropriately, ie, requests for full details of impurities to be included on early phase clinical specifications. However, it is difficult to envisage the compilation of a specification for any early phase regulatory submission, .e.g. investigational Medicinal Product Dossier (IMPD), investigational new drug (IND), without applying some aspects of the ICH Q6A guidance. Nonetheless, great care is needed to ensure that specifications are not constrained at an early stage based on limited data, especially if there is an intention to remove tests specified in early versions.
ICH Q6A does not include any discussions on process capability (Cp). However, it has become clear with the advent of QbD that a comprehensive understanding of the effect of variability of the process and supporting methodologies is required before robust specifications can be set. Process capability measures the output of an ‘in-control’ process by assessing the ratio of the process specification width (or range) to the spread of process values using standard deviation units. As the process capability improves, the variability decreases, and it becomes more closely centred between the specification limits. A process where the majority of all measurements reside within the allowable specification range is a 6-sigma capable process3 (Table 1).
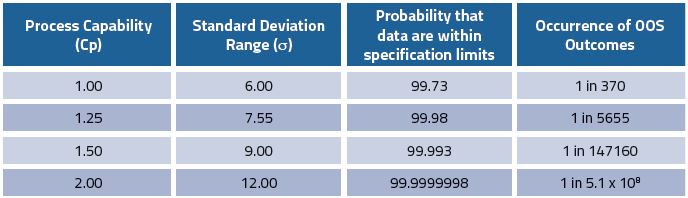
Table 1: Process capability (Cp) and the likelihood of out of specification (OOS) consequences (adapted from Elder3)
This assumes that the data are centred within the specification range, ie. 100% for API potency, ± 2% for specified range, ie. 98.0-102.0%. However, this is not typically the case; the presence of impurities means that the drug substance process is off-centred at 100-x%, where x is the total value of all impurities found in the process. This is reflected by the off-target process capability index (Cpk). A process with a Cp of 2 could have a Cpk of 1.5, which would equate to a failure rate of four defects in every million assessments. Thus, logically process capability or variability (including method variability), is one of the key parameters in setting robust specifications.
However, regulators have indicated that it is “not considered appropriate to add method variability as determined in analytical method validation to the variation seen in batch results as this variability is already included in the batch results and therefore will be counted twice” .10 This perspective is predicated on the view that measurement uncertainty will always be smaller than batch variation. This is true if the sample size of the number of batches in question are sufficiently large reflecting the total population (typically a minimum of 30 batches).11 In practice many new drug products will submit regulatory files with a limited number of batches, ie. three at commercial scale, accompanied by a number of development batches at smaller scale. This prompts the key question: “How will it be possible to ensure the variability of the production process is covered by the specification with limited number of manufacturing scale lots that are available at the time of submission?”3 Are these criteria replicated by the product’s clinical experience or are the limits based on the cumulative process performance, or both?”10
Thus, if the specification is tightened during the review process and / or takes no account of process / method variability, this can turn a capable process into a non-capable process leading to OOS results or batch failures. Unfortunately, this is an increasingly common incidence during regulatory review, where reviewers consider that by further constraining the proposed specification they will make the process / specification more discriminating ensuring patient safety; simply, all that occurs are more batch failures of product that are of an acceptable quality.3
It is widely accepted that method variability is frequently greater than manufacturing process variability, particularly for API processes.12-13 Intermediate precision is the most appropriate method validation parameter for assessing Cp and should be taken into consideration when proposing any specification limits, or when assessing the capability of the method when the specifications are “constrained”, as is the case for API assay, ie. 98.0-102.0%. Therefore, a specification of 100.0 ±2% i.e. 4% range for a 3-sigma process is equivalent to a total variability of 0.67%.Thus the method variability needs to be at least half this value, ie. 0.34% (or less).13 The allowable method variability is further constrained as the true means of the specification is less than 100%, ie. 100- total impurities.
Thus, the supporting methods need to be 6σ capability (ie. show decreased variability) and hence the total method deviation is required to be ≤ one twelfth of the total allowable range or tolerance, ie. 0.17% (or less).13 Based on this significant high-performance liquid chromatography (HPLC) method variability, several commentators14-16 have expressed significant misgivings about the utility of the standard HPLC assay method to monitor drug substance quality. Kredla et al.17 commented that, “assay results are simply not stability-indicating [….] due to the large assay variability associated with them”.
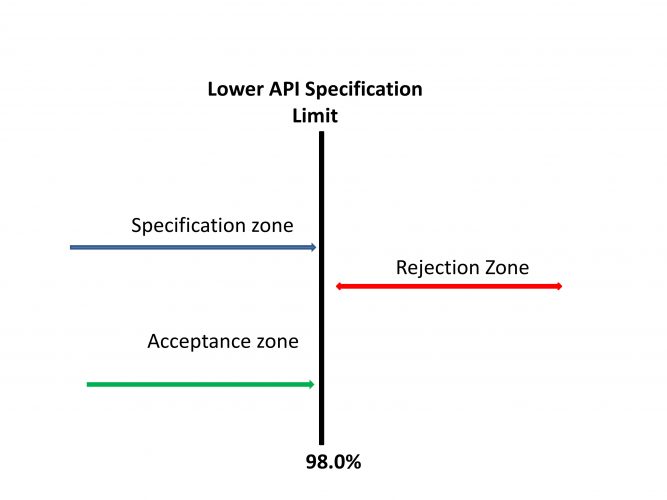
Figure 1: Specifications Complying with Simple Acceptance Criteria. Adapted from Ellison and Williams18
In the classical operating scenario, specifications have clearly defined acceptance and rejection zones (Figure 1).
The various pharmacopoeias are currently evaluating applying decision rules based on a probabilistic assessment that acknowledges measurement uncertainty (MU) and the role it plays in decision making, ie. Acceptance / rejection, based on compliance with a pre-determined specification.19 These decision rules support the decision-making process by, (i) assessing the measurement result, (ii) the specification limit (both current practices), (iii) the measurement uncertainty, (iv) and assessing the acceptable level of probability of making a wrong decision (where (iii) and (iv) are new approaches). These specification decision rules will introduce the concept of a “guard band” or “transition zone” between the acceptance and rejection zones within a specification.3 (Figure 2).
Within the specification “transition zone” a product will be considered non-compliant if the probability of either being above or below the designated upper or lower specification limit exceeds 2.5%. A target MU equivalent to σ equal to 1.02 would meet these requirements. However, a bias will impact on this acceptance value. In the case of a non-centred specification or method bias of 1.0%, then σ needs to be reduced to 0.6%.3
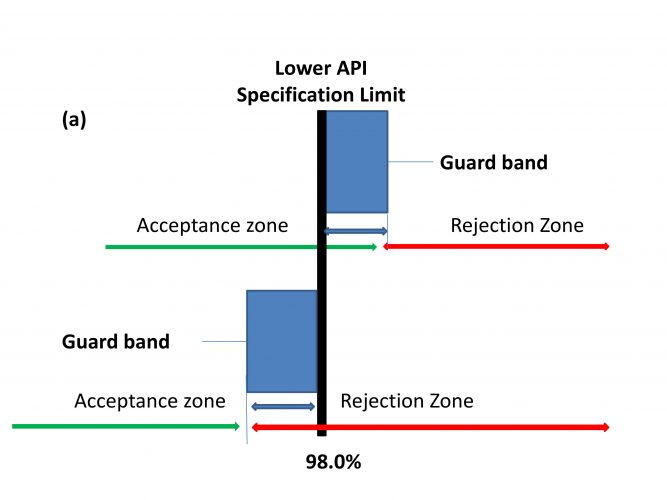
Figure 2: Specifications using Acceptance Criteria, (a) Relaxed acceptance (test for non-conformity), (b) Stringent acceptance (test for conformity). Adapted from Ellison and Williams18
An increasingly common situation that can occur in the QbD paradigm is the setting of specifications based solely on the clinically derived CQAs, without considering Cp arguments where Cp assessments clearly indicate that these proposed specification changes will make this a non-capable process, ie. a 3-sigma process. The recent trend towards tightening dissolution specifications from the typically encountered Q=80% at 30 minutes, to the much tighter Q=80% at 20 minutes is a case in point. The intrinsically poor hydrodynamics of the dissolution apparatus at time points below 30 minutes ensures that there is significantly increased method variability for methods based on 20-minute (or lower) time points.20 That is, the method becomes less robust, but not more discriminating.
A potential solution is to move towards the clinically-derived specification in managed stages, ie. post-approval. Applicants would therefore generate data from a meaningful number of batches, ie, 30 batches; allowing an assessment of the population variability and adjust the specification based on a revised Cp assessment.21 However, for some low volume products these large numbers of manufactured batches, i.e. ≥30 are still very aspirational.
References
ICH Q6A. 1999. SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR NEW DRUG SUBSTANCES AND NEW DRUG PRODUCTS: CHEMICAL SUBSTANCES. Current Step 4 version dated 6 October 1999.
ICH Q8(R2). 2009. PHARMACEUTICAL DEVELOPMENT. Current Step 4 version dated August 2009.
Elder DP. 2018. ICH Q6A specifications In ICH Quality Guidelines, An Implementational Guide, Eds. Teasdale A, Elder DP and Nimms R, Wiley J, pp. 433-466.
ICH Q9. 2005. QUALITY RISK MANAGEMENT. Current Step 4 version dated 9 November 2005.
ICH Q10. 2008. PHARMACEUTICAL QUALITY SYSTEM. Current Step 4 version dated 4 June 2008.
ICH M7(R1). 2017. ASSESSMENT AND CONTROL OF DNA REACTIVE (MUTAGENIC) IMPURITIES IN PHARMACEUTICALS TO LIMIT POTENTIAL CARCINOGENIC RISK. Current Step 4 version dated 31 March 2017.
ICH M3(R2). 2009. GUIDANCE ON NONCLINICAL SAFETY STUDIES FOR THE
CONDUCT OF HUMAN CLINICAL TRIALS AND MARKETING AUTHORIZATION FOR PHARMACEUTICALS . Current Step 4 version dated 11 June 2009.
ICH Q3A(R2). 2006. IMPURITIES IN NEW DRUG SUBSTANCES. Current Step 4 version dated 25 October 2006.
ICH Q3B(R2). 2006. IMPURITIES IN NEW DRUG PRODUCTS. Current Step 4 version dated 2 June 2006.
EMA. 2011. Report on the expert workshop on setting specifications for biotech products, European Medicines Agency, London, 09 September, 2011. EMA/CHMP/30584/2012.
Porter WR. 2012. Impact of stability on setting and meeting specifications in an uncertain world, MSBW 2012. http://www.mbswonline.com/upload/presentation_5-25-2012-10-43-30.ppt. Accessed on 17th August 2018.
Tsang PKS, Larew JSA, et al. 1998. Statistical approaches to determine analytical variability and specifications: Application of experimental design and variance component analysis. J. Pharm. Biomed. Anal., 16, 1125-1141.
Dejaegher B, Jimidar M et al. 2006. Improving method capability of a drug substance HPLC assay. J. Pharm. Biomed. Anal., 42, 155-170.
Ermer J. 2001. Validation in pharmaceutical analysis. Part I: An integrated approach. J. Pharm. Biomed. Anal., 24, 755-767.
Hofer JD, Olsen BA, et al. 2007. Is HPLC assay for drug substance a useful quality control attribute? J. Pharm. Biomed. Anal., 44, 2007; 906-913.
Ermer J. In: Ermer J, Miller JHMcB Eds). 2005. Method validation in pharmaceutical analysis. A guide to best practice. Wiley-VCH GmbH and Co., KGaA, Weinheim, p. 88.
Skrdla PJ, Wang T, et al. 2009. Use of a quality-by-design approach to justify the removal of the HPLC weight % assay from routine API stability testing protocols. J. Pharm. Biomed. Anal., 50, 794-796.
Ellison SLR, Williams A (Eds). 2007. Eurachem/CITAC guide: Use of uncertainty information in compliance assessment. http://www.eurachem. org. Accessed on 17th August 2018.
Weitzel J. 2014. Lifecycle Management of Analytical Procedures: What is it all about? http://www.aaps.org/uploadedFiles/Content/Sections_and_Groups/Regional_Discussion_Groups/Southern_California_Pharmaceutical/Resources/Applying%20Lifecycle%20Approach%20to%20a%20Method%20rev%202.pdf. Accessed on 17th August 2018.
Grady H, Elder DP, et al. 2018. Industry’s View on Using Quality Control, Biorelevant, and Clinically Relevant Dissolution Tests for Pharmaceutical Development, Registration, and Commercialization. J. Pharm. Sci., 107, 34-41.
Moran E, Perry M. 2011. Setting specifications, statistical considerations. EuropaBio Meeting. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/10/WC500115818.pdf. Accessed on 17th August 2018.
Biography
DAVE ELDER has nearly 40 years of service within the pharmaceutical industry at Sterling, Syntext and GlaxoSmithKline. He is now an independent GMC consultant. Dr Elder is a visiting professor at King’s College, London, and is a member of the British Pharmacopoeia. He is a member of the Joint Pharmaceutical Analysis Group (JPAG), and a member of the Analytical Division Council of the Royal Society of Chemistry.
The rest of this content is restricted - login or subscribe free to access
 Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Why subscribe? Join our growing community of thousands of industry professionals and gain access to:
- bi-monthly issues in print and/or digital format
- case studies, whitepapers, webinars and industry-leading content
- breaking news and features
- our extensive online archive of thousands of articles and years of past issues
- ...And it's all free!
Click here to Subscribe today Login here