

Thermodynamics and kinetics driving quality in drug discovery
Posted: 31 August 2011 |
Recently, there has been renewed interest in using thermodynamic and kinetic data, alongside empirical rules (particularly focused upon cLogP and molecular weight) and guiding metrics such as ligand efficiency and lipophilic ligand efficiency developed for fragments, leads and drugs in order to facilitate the design of compounds with a greater chance of producing successful drugs1. This interest has been assisted both by improvements in instrumentation as well as evidence that thermodynamically and kinetically optimised compounds fare better in the clinic2.
Optimisation of the binding affinity, which may have to be improved by several orders of magnitude from initial hit to drug molecule, can be achieved by modifying the individual thermodynamic and kinetic contributions. However, medicinal chemists have, up to now, been reluctant to consider these measurements during hit selection and lead optimisation, because it has been difficult to understand how the different design strategies affect the individual forces resulting in different thermodynamic and kinetic profiles. By incorporating both retrospective analysis and real time data collection in active projects, the value of using these fundamental contributions to guide the selection of chemical start points and how they can be used to influence optimisation strategies will become clear.
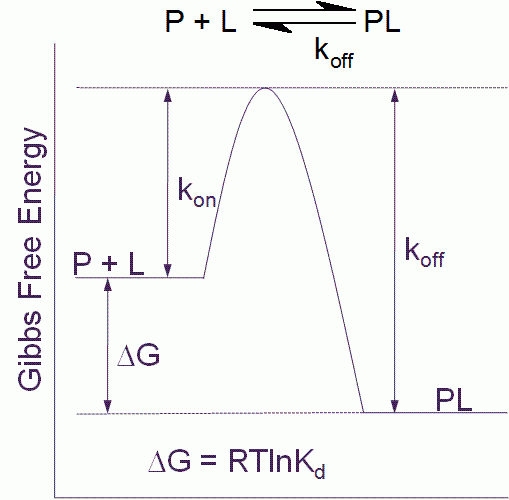
Figure 1 Free energy diagram for a single step binding interaction between protein (P) and ligand (L). For a binding reaction of this type increasing affinity is achieved by lowering the free energy of the PL complex. Increasing residence (decreasing koff) time is achieved by lowering the free energy of the PL complex and/or destabilizing the transition state
Recently, there has been renewed interest in using thermodynamic and kinetic data, alongside empirical rules (particularly focused upon cLogP and molecular weight) and guiding metrics such as ligand efficiency and lipophilic ligand efficiency developed for fragments, leads and drugs in order to facilitate the design of compounds with a greater chance of producing successful drugs1. This interest has been assisted both by improvements in instrumentation as well as evidence that thermodynamically and kinetically optimised compounds fare better in the clinic2.
Optimisation of the binding affinity, which may have to be improved by several orders of magnitude from initial hit to drug molecule, can be achieved by modifying the individual thermodynamic and kinetic contributions. However, medicinal chemists have, up to now, been reluctant to consider these measurements during hit selection and lead optimisation, because it has been difficult to understand how the different design strategies affect the individual forces resulting in different thermodynamic and kinetic profiles. By incorporating both retrospective analysis and real time data collection in active projects, the value of using these fundamental contributions to guide the selection of chemical start points and how they can be used to influence optimisation strategies will become clear.
Binding affinity
The binding affinity or Kd for a simple one step binding reaction between protein and ligand, as shown in Figure 1, can be resolved into both thermodynamic and kinetic contributions:
Kd = e(DG/RT) = e(DH/RT – DS/R) = koff/kon
where ΔG is the Gibbs free energy of binding, ΔH is the enthalpy of binding, ΔS is the entropy of binding , R is the gas constant, T the temperature in Kelvin and kon and koff are the association and dissociation rate constants respectively.

Figure 1 Free energy diagram for a single step binding interaction between protein (P) and ligand (L). For a binding reaction of this type increasing affinity is achieved by lowering the free energy of the PL complex. Increasing residence (decreasing koff) time is achieved by lowering the free energy of the PL complex and/or destabilizing the transition state
It is important to point out that the kinetic and thermodynamic parameters, whilst they relate to the same binding interaction, contribute differently to our understanding of the process. Thermodynamics tells us about the spontaneity of the binding interaction, but nothing about the time dimension of the process. Kinetics on the other hand tells us how fast the reaction will proceed, but provides no information about the final state.
Thus, it is clear that for a full understanding of the binding interaction we need to understand both the factors that contribute to the thermodynamics and also the kinetics that together control the binding reaction.
The complex array of fundamental forces that underlie the binding interaction and dictate the measured thermodynamic and kinetic parameters can be simplistically classified as non-specific or specific. This takes into account both those forces that occur within the binding site and also those important forces operating outside of the binding site, which together control the interaction of proteins and ligands.
Non-specific forces tend to be long range forces that decay proportionally with the distance between the interactants. They include the electrostatic double-layer force, van der Waals force and steric forces3.
These forces can be attractive or repulsive, depending upon the polarisability of the atoms involved in the binding interaction, the surface charge density and hence the counter ion concentration near the surface of charged partners, the geometry of the interactants and also the media in which the interaction takes place.
Specific interactions include those noncovalent interactions whose highly complementary nature, both in shape and chemical nature, are typified in molecular recognition. These are the multiple short range interactions, including van der Waals, hydrogen bonds and hydrophobic contacts that together serve to counteract any non-specific repulsive forces, and contribute predominantly to the free energy of binding.
It is these short range specific forces that can potentially be engineered in order to improve the binding affinity, with maximal affinity achieved when the most favourable con – tribution from each of these sources can be accessed4.
Isothermal Titration Calorimetry (ITC) and thermodynamics
ITC is the only biophysical technique that can measure the binding free energy and total binding enthalpy directly in a single experiment. The total binding entropy is then calculated from the equation:
DS = (DH-DG)/T
with the binding experiment also providing the stoichiometry of the interaction. Thus, a single ITC experiment provides a full thermodynamic characterisation of the binding interaction, and has traditionally been used to help to understand the different contributions to affinity5.
However, the introduction of new instruments providing reduced sample consumption and automated operation has now allowed ITC to be implemented in a proactive manner, where studies during hit and lead selection and optimisation may impact on compound design. The ability to investigate up to 48 binding interactions per day, allowing many more representatives of discrete chemical series to be tested, coupled with initial suggestions that improved enthalpy may correlate with improved physicochemical properties and potentially better efficacy has intrigued medicinal chemists. Using ITC to deconstruct the binding affinity of hits and leads into their component thermodynamic signatures is essential in trying to understand and modify the contributions from the different types of forces involved in binding. Initial hits with favourable enthalpies may be selected and the optimisation process monitored using ITC in order to obtain a balanced optimisation, whereby the inflation of molecular weight and lipophilicity, typically associated with traditional lead optimisation strategies, may be avoided or mitigated. Balanced optimisation would result from modifications in thermodynamic properties such that every 10 fold increase in binding affinity (ΔΔG of -6 kJ/mol,) at 37°C, would be achieved by a favourable enthalpy change of around -3 kJ/mol and a favourable entropy change of around 10 J/mol.K. These values therefore place some thermodynamic guidelines on the changes in hydrophobic interactions, potentially leading to entropic gains, and van der Waals and hydrogen bonding interactions typically associated with favourable enthalpy gains.
As well as providing a check on the progression of a balanced optimisation approach, monitoring of the lead optimisation process by ITC offers the opportunity to identify potential reasons for the lack of affinity gains that may occur. For example, even after the introduction of a specific modification to a lead compound, designed to improve affinity, compensating changes in either enthalpy or entropy can reduce the effect of the modification. The thermodynamic signature provided by ITC can provide a means of identifying the causes of enthalpy-entropy compensation6 and offer potential approaches to overcome this common phenomenon.
Surface Plasmon Resonance (SPR) and kinetics
The measurement of the kinetics of proteinligand interactions has been facilitated enormously by the use of SPR-based approaches. These methods make use of the reduction in intensity of reflected light brought about by a change in critical angle due to the resonant energy transfer between an evanescent wave and the surface plasmons in the gold layer of the biosensor. These resonance conditions are influenced by the mass change at the surface. Typically, the protein of interest is coupled to the sensor surface, with the compounds of interest flowed sequentially over the immobilised protein in a direct binding assay7.
As with ITC, recent improvements in instrumentation leading to higher throughput, faster data collection rates and improved sensitivity have allowed SPR measurements to have a wider impact in drug discovery. Throughput of up to 4800 interactions analysed in 24 hours has enabled the approach to be used both for fragment screening, as a hit evaluation tool post traditional high throughput screening, and more conventionally to characterise detailed binding kinetics. This versatility has recently been coupled with renewed interest in measuring drug-target residence times in order to improve efficacy8.
The residence time (τ) for the simple one step binding interaction shown in Figure 1 is the reciprocal of the dissociation rate constant:
t = 1/koff
but becomes more complicated for binding mechanisms involving conformational changes.
However, residence times cannot be considered unless association occurs in the first place, and the observed rate constant for association (kobs) is dependent upon the association rate constant as well as the concentration of ligand, according to the relationship:
kobs = kon.[L] + koff
with more complex expressions required for mechanisms involving conformational changes.
SPR potentially allows those compounds with characteristic slow association or dissociation kinetics, where t½ may be of the order of minutes to hours, to be fully evaluated, when they would potentially show poor behaviour in most binding assays at a fixed time.
Investigation of the binding kinetics, not only the thermodynamics of the binding interaction, allows a more thorough examination of the structure activity relationships (SAR) influencing the pharmacokinetic and pharmacodynamic profiles of lead compounds, alongside traditional measured parameters such as adsorption, distribution, metabolism and excretion (ADME).
For example, slow binding compounds will require higher free drug levels, established by increased dose, in order to attain the required target occupancy, whereas long target residence times have been advocated as improving duration of action allowing improved dosing regimens, improved selectivity and reduced toxicity by providing a greater therapeutic index.
Improving compound quality
Recently, greater emphasis has been placed on improving the quality of lead compounds, rather than increasing binding affinity alone during the hit to lead process. Several efficiency indices (see Table 1) have been described in the literature which can be used as tools to facilitate the selection and further optimisation of compounds which may offer greater probability of later stage success.
Table 1: Ligand efficiency and other efficiency metrics
Index | Calculation | Reference |
Ligand Efficiency (LE) | DG/NHA | 9 |
Lipophilic Ligand Efficiency (LLE) | pKi-LogD | 10 |
LogP/Ligand Efficiency (LELP) | LogP/LE | 11 |
Enthalpic Efficiency (EE) | DH/NHA | 12 |
Kinetic Efficiency (KE) | t/NHA | 13 |
The first of these efficiency metrics to be proposed was Ligand Efficiency (LE), which attempted to relate the binding affinity to the size of the compound giving a so-called ‘bang for your buck’ measure of the effectiveness of increasing potency by growing fragments or compounds. Lipohilic Ligand Efficiency (LLE) is an alternative to LE, which takes into account lipophilicity and allows guidance on the amount of specific binding achieved for a specific value of LogD. Ligand Efficiency / LogP (LELP) can be considered as a composite metric attempting to relate the efficiency of binding with the lipoophilicity of the ligand. All of these metrics are useful in helping to guide medicinal chemistry design by allowing optimisation towards ideal values for the given metric (for a good hit or lead values are LE > 0.4, LLE > 7, and 0 < LELP < 7.5). However, they all rely upon measurements of ΔG or the equivalent apparent equilibrium value. However, Enthalpic Efficiency (EE) and Kinetic Efficiency (KE) offer additional information relating to the thermodynamic and kinetic signatures that may be desired of lead compounds, based upon the favourable forces of binding, as described above. EE values can be used most effectively to discriminate between compounds within chemical series, as this metric will quantify the compound’s ability to form non-covalent bonding interactions on a per heavy atom basis. Using the metric in this way will tend to circumvent problems arising from changes in binding mode or mechanism, which may tend to be associated with different net changes in the overall bonding inventory, which will be more difficult to rationalise.
By monitoring KE alongside these other metrics, the medicinal chemist will be able to incorporate strategies focused upon changes in residence time per heavy atom, designed to either increase or decrease the observed residence time, depending upon whether a long or short duration of action at the enzyme or receptor is required. This will facilitate more rational changes in lipophilicity, polarity, molecular size, and bonding interactions aimed at producing the desired kinetic and thermodynamic effects.
Summary
The binding affinity can be broken down into thermodynamic (enthalpy and entropy) and kinetic (rate of association and dissociation) components. Previously, optimisation of binding affinity has been undertaken with little regard for the factors that influence these contributions, partly through a lack of good quality data, and partly through a lack of understanding of chemical strategies that may modify the forces responsible for the magnitude of these contributions. ITC is the only technique that allows a direct measurement of the binding enthalpy. With recent improvements in instrumentation, allowing lower reagent consumption and higher throughput, thermodynamic characterisation on a wide number of lead candidates is now a realistic possibility. Methods to characterise the kinetics of inhibition from pre-steady state assays are well understood, but have suffered from a lack of throughput and impact on lead optimisation. SPR approaches to measure rate constants are now established in most biophysical laboratories, facilitating kinetic measurements on a larger scale, creating the opportunity for kinetic data to gain in prominence.
Together, thermodynamic and kinetic studies on hits and leads will improve our fundamental understanding of protein-ligand interactions and will enable new and improved strategies for successful drug discovery.
References
1. Smith, G.F (2009) Medicinal Chemistry by the Numbers: The Physicochemistry, Thermodynamics and Kinetics of Modern Drug Design. Prog. Med. Chem. 48, 1 – 29
2. Freire, E (2008) Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 13, 869 – 874
3. Leckband, D (2001) Measuring the forces that control protein interactions. Annu. Rev. Biophys. Biomol. Struct. 29, 1 -26
4. Freire, E (2009) A thermodynamic approach to the affinity optimization of drug candidates. Chem. Biol. Dug Des. 74, 468 – 472
5. Ward, W.H and Holdgate, G.A (2001). Isothermal titration calorimetry in drug discovery. Prog. Med. Chem. 38, 309 – 376
6. Starikov, H.B and Norden, B. (2007) Enthalpyentropy compensation: a phantom or something useful? J. Phys. Chem. B 111, 14431 – 14435
7. Huber, W (2005) A new strategy for improved secondary screening and lead optimization using high-resolution SPR characterization of compound–target interactions. J. Mol. Recognit. 18, 273 – 281
8. Lu, H. and Tonge, P.J. (2010) Drug-target residence time: critical information for lead optimization. Curr. Opin. Chem. Biol. 14, 467- 474
9. Hopkins, A.L. et al (2004) Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 9, 430 – 431
10. Leeson, P.D. and Springthorpe, B. (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug. Discov. 6, 881-890
11. Keseru, G.M. and Makara, G.M. (2009) The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug. Discov. 8, 203 – 212
12. Ladbury, J.E. (2007) Enthalpic efficiency and the role of thermodynamic data in drug development: possibility or a pipeline dream! European Pharmaceutical Review 6. (http://www.european pharmaceuticalreview.com/)
13. Holdgate, G.A. and Gill, A.L. (2011) Kinetic Efficiency: the missing metric for enhancing compound quality DDT (in press)
Geoff Holdgate gained his BSc (Hons) in Biological & Biochemical Sciences from the University of Salford in 1992. He then joined ICI Pharmaceuticals (now AstraZeneca), as a research scientist working in Molecular Enzymology. He was appointed Principal Scientist in Enzymology and Biophysics in 2006, and has worked on three marketed medicines.
