Native liquid chromatography coupled to mass spectrometric analysis for the assessment of higher order structures of proteins
Posted: 28 April 2020 | Dr Ioannis A Papayannopoulos (Celldex Therapeutics) | No comments yet
The accurate measurement of the molecular mass of proteins is an essential component of protein characterisation. In this article, Ioannis Papayannopoulos discusses the applications of native protein chromatography coupled to mass spectrometry to enable the characterisation of tertiary structures and non-covalent interactions.
LIQUID CHROMATOGRAPHY coupled to electrospray mass spectrometry (LC-MS) has been useful in the analysis of intact proteins or large subunits thereof. Important molecular mass information helps to establish and assess identity, purity and homogeneity of proteins.1 In addition, extensive use, over four decades, of reversed phase (RP)-LC-MS has enabled the separation and analysis of peptides, generated from enzymatic digestion of proteins, in order to confirm amino acid sequences and to characterise amino acid modifications.2 However, the organic solvents used for elution of analytes and the strong acids used as ion pairing agents, necessary for performing RP-HPLC separations, while highly compatible with mass spectrometry usually result in denaturation of proteins and the dissociation of protein complexes and therefore the loss of higher order structural information.
The accurate measurement of the molecular mass of proteins, separated and purified by RP-HPLC, is an essential component of protein characterisation”
Chromatographic separations of proteins under conditions and with solvents that do not cause denaturation, chiefly size exclusion and ion exchange, have also been used extensively for protein characterisation.3,4 However, the non-volatile salts in the mobile phase of such separations, while necessary for maintaining proper folding and non-covalent interactions, have been incompatible with LC-MS due to the suppression of ionisation of protein analytes and the formation of salt adducts of proteins that make data interpretation difficult. Recent advances in chromatographic separations using novel column chemistries, which yield good separation of analytes with buffers that contain volatile salts compatible with mass spectrometry, facilitate the analysis by LC-MS of proteins and of heterogenous macromolecular complexes in their native state.5,6 The use of ion mobility mass spectrometry provides additional insights into the native structures of these species.7,8
Denaturing LC-MS of proteins
Efficient analysis of intact proteins by RP-LC-MS has been a more recent development compared to the analysis by RP-LC-MS of peptides obtained from enzymatic digestion of proteins. Peptide RP-LC-MS has been an essential component of the characterisation of proteins, by matching experimental data to known amino acid sequences; it is also the enabling technology in proteomics for the identification of unknown proteins by using such data to search protein databases. Protein RP-LC-MS provides an assessment of the whole molecule: the measured mass will either match the mass calculated from the amino acid sequence, indicating that there are no unexpected modifications, amino acid substitutions or clips; or the measured mass will not match the calculated mass, in which case the difference between measured and calculated mass often provides valuable insights on possible post-translational and other modifications to the sequence. However, non-covalent protein-protein, protein-small molecule and protein-DNA/ RNA complexes generally dissociate under the denaturing conditions of RP-HPLC and cannot be measured.
Non-denaturing LC-MS of proteins
Size exclusion chromatography (SEC) and ion exchange chromatography (IEC) are typically non-denaturing methods that can separate proteins by size (SEC) or by charge (IEC). Both types of separation are carried out with mobile phases that introduce minimal or no disruption of intramolecular and intermolecular hydrogen bonds, salt bridges and hydrophobic interactions, thus preserving higher order structure and protein complexes. These modes of native liquid chromatography coupled with ultraviolet (UV) detection of analytes are used in the analysis and characterisation of protein therapeutics to detect and quantitate critical quality attributes such as aggregates (SEC) or charge variants (IEC). They are also used in many research areas to identify and characterise protein complexes as well as non-covalent complexes between proteins and DNA/RNA or proteins and small molecules. However, their coupling with on-line mass spectrometry detection and measurement has been problematic because the buffers used to ensure that the protein samples maintain their native state during analysis typically contain non-volatile sodium, potassium, phosphate and perchlorate salts.
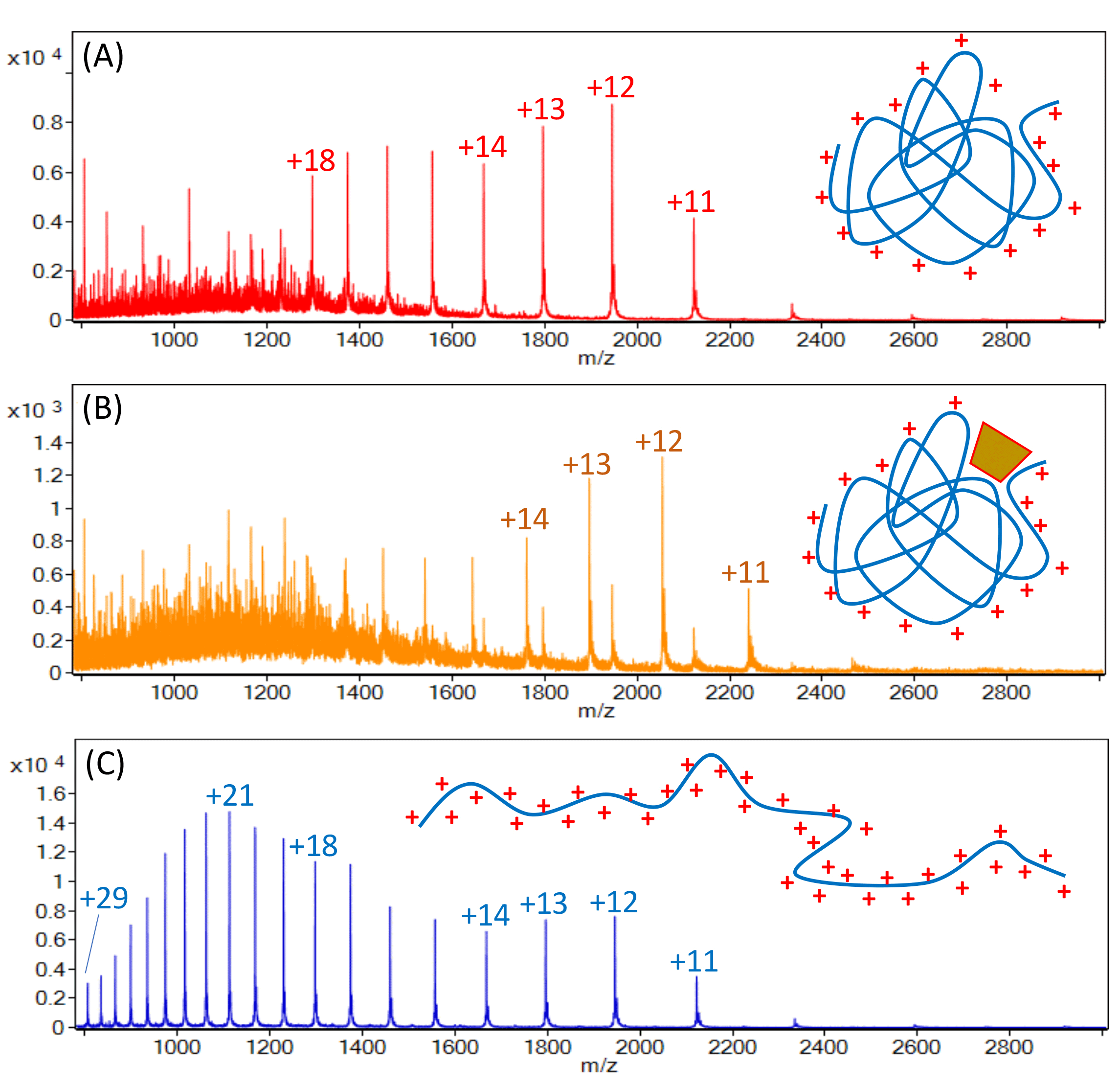
Figure 1: A small (24.5kDa) protein analysed by SEC-MS under native conditions (A) and under native conditions with a non-covalently bound 1.5kDa ligand (B). The complex dissociates completely under denaturing RP-HPLC conditions (C). It is worth noting that the native state folded protein exposes less of its surface to the solvent and yields ions with fewer charges (higher m/z) than the denatured and unfolded protein.
Efforts to substitute solvents with ones that are compatible with mass-spectrometry by containing volatile salts (eg, ammonium formate or ammonium acetate) and not containing other components (eg, detergents) that adversely affect ionisation and measurement of non-denatured proteins or protein complexes, have been made for some time, with limited success. In recent years, however, improvements in chromatographic columns have yielded good separation of native state protein samples with elution conditions that are compatible with mass spectrometry. Thus, it has become possible, for example, to directly measure the interaction of a folded protein with a small molecule that is bound to it non-covalently (Figure 1).
Ion exchange chromatography (IEC) of proteins is carried out by loading the protein onto the IEC column in a low ionic strength buffer and eluting the various charged species by gradually increasing the ionic strength of the elution buffer. The development of stationary phases that provide adequate retention and resolution of the analytes with elution solvents that contain volatile salts and are thus compatible with mass spectrometry, has enabled direct measurement of the masses of protein charge variants, which cannot be separated under the denaturing conditions of RP-HPLC (Figure 2).
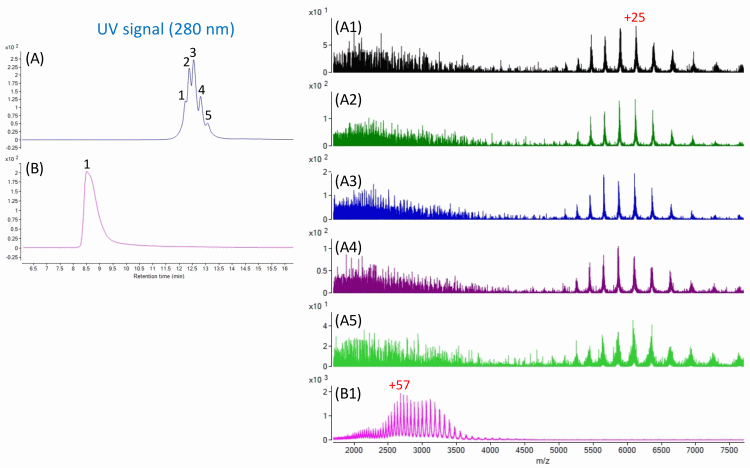
Figure 2: Charge variants of an antibody molecule separated by IEC-HPLC (A) and measured by mass spectrometry (A1-A5), using an IEC column optimised to perform well with compatible HPLC solvents compatible with mass spectrometry. These all coelute under RP-HPLC conditions (B) and yield a single mass spectrum (B1), which comprises multiple charged ions from all the charge variants. The relative abundance of the different species can be readily determined from the UV signal of the elution profile. It is worth noting that the native IEC-analysed antibody yields lower charge states (ie, fewer charges on the molecule) than the denatured RP-analysed antibody.
Ion mobility mass spectrometry
Ions in the gas phase can be separated based on their shape, charge and mass by interaction with a buffer gas; this is the principle of ion mobility spectrometry. It is a process that occurs on a millisecond time scale and so can be readily coupled with HPLC separations, which occur on a time scale of seconds, and with mass spectrometry in which mass-to-charge (m/z) separations and measurements take place on a microsecond time scale. Thus, the chromatographic separation of proteins, whether based on hydrophobicity (RP-HPLC), size (SEC-HPLC) or charge (IEC-HPLC), can be further enhanced by ion mobility separations based on shape, before mass measurements are carried out with the mass spectrometer (Figure 3).
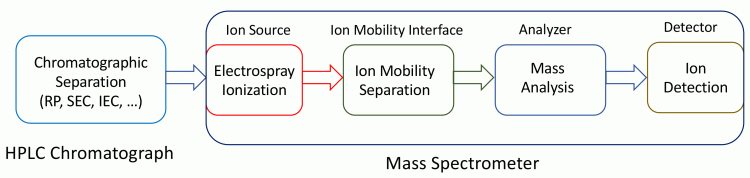
Figure 3: HPLC coupled with a mass spectrometer that is equipped with an ion mobility module.
In this manner, ionised molecules of the same mass but with different shapes (eg, proteins folded differently such as IgG2 isoforms or non-covalent complexes with different arrangements of the same protein subunits) can be separated in the gas phase before each is analysed with the mass spectrometer. The rotationally averaged collision cross-section (CCS) of a molecule encompasses information about the shape of the ionised molecule in the gas phase and when measured under standardised conditions can be independent of specific instruments. Thus, CCS values are very useful in comparing experimental data involving the higher order structure of proteins that are separated by non-denaturing chromatographic methods, which preserve native protein structures.
Conclusion
Ion mobility provides an additional mode of separation based on the shape of gas phase protein ions”
The accurate measurement of the molecular mass of proteins, separated and purified by RP-HPLC, is an essential component of protein characterisation, used to confirm that a known amino acid sequence is correct or to identify potential modifications. The loss of the higher order structure of proteins due to the denaturing effect of the RP-HPLC solvents can be addressed by the use of complementary separation methods such as size exclusion and ion exchange HPLC, which are carried out under conditions that preserve higher order structures due to protein folding and non-covalent adducts, aggregates and complexes. These non-denaturing HPLC separations can be coupled with mass spectrometry, thus allowing direct mass measurements of these native forms. Ion mobility provides an additional mode of separation based on the shape of gas phase protein ions.
About the author
Dr Ioannis A Papayannopoulos has been carrying out peptide and protein analytical work for the past several decades, using mass spectrometry, chromatography, spectroscopy and other analytical techniques. He received his undergraduate degree in chemistry from Bowdoin College and his PhD in Organic Chemistry from the Massachusetts Institute of Technology under the supervision of the late professor Klaus Biemann. Over the years Ioannis has alternated between academia, most recently as director of the Proteomics Facility at the Koch Institute for Integrative Cancer Research at MIT, and the biopharmaceutical industry in senior scientific and management roles at companies such as Biogen, Covance and AstraZeneca. For the past five years he has been with Celldex Therapeutics working on the analysis and characterisation of antibodies, antibodydrug conjugates and recombinant protein pharmaceuticals.
References
- van de Merbel NC. Bioanalysis 11, 629-644 (2019).
- Miao Q, et al. Mass Spectrometry Reviews 36, 1-20 (2016).
- Ardelt W, Laskowski Jr M. Analytical Biochemistry 120, 198-203 (1982).
- Hong P, Koza S, Bouvier P. Journal of Liquid Chromatography and Related Technologies 35, 2923-2950 (2012).
- Bailey AO, et al. MABS 10, 1214-1225 (2018).
- Haberger M, et al. MABS 8, 331-339 (2016).
- Marcoux J, et al. Protein Science 24, 1210-1223 (2015).
- Lanucara F, et al. Nature Chemistry 8, 331-339 (2014).
Issue
Related topics
Analytical techniques, Chromatography, Liquid Chromatography - Mass Spectrometry (LC-MS), Mass Spectrometry, QA/QC