

Comparison of new vaccine approaches – COVID-19
Posted: 26 June 2020 | John Liddell (CPI) | 2 comments
The promise of virally vectored DNA and mRNA vaccines is tremendous in terms of vaccine safety and speed of response to new pathogens. This article explores why these approaches have attracted attention in light of the COVID-19 pandemic and why they are highly appropriate for rapid vaccine development and deployment.
THE COVID-19 pandemic has raised the profile of prophylactic technology and increased awareness of the challenges associated with developing vaccines rapidly. While vaccinology has a long history, now is an appropriate time to review promising technologies that have taken centre stage in the race to develop a vaccine for SARS-CoV-2 and that illustrate the technical convergence from the vaccine and gene therapy fields, which are both benefitting from research work in synthetic and cell biology.
As of 20 April 2020, there were 76 vaccine candidates in development for COVID-19 according to the World Health Organization (WHO). The chart in Figure 1 shows the breakdown by vaccine type. Two sectors – viral vectors and RNA vaccines – are among the newest approaches for vaccine development at the forefront of COVID-19 vaccine trials. This review will focus on why these platforms have attracted attention and why they are highly appropriate for rapid vaccine development and deployment. Technical approaches established for gene therapy applications are also explored.
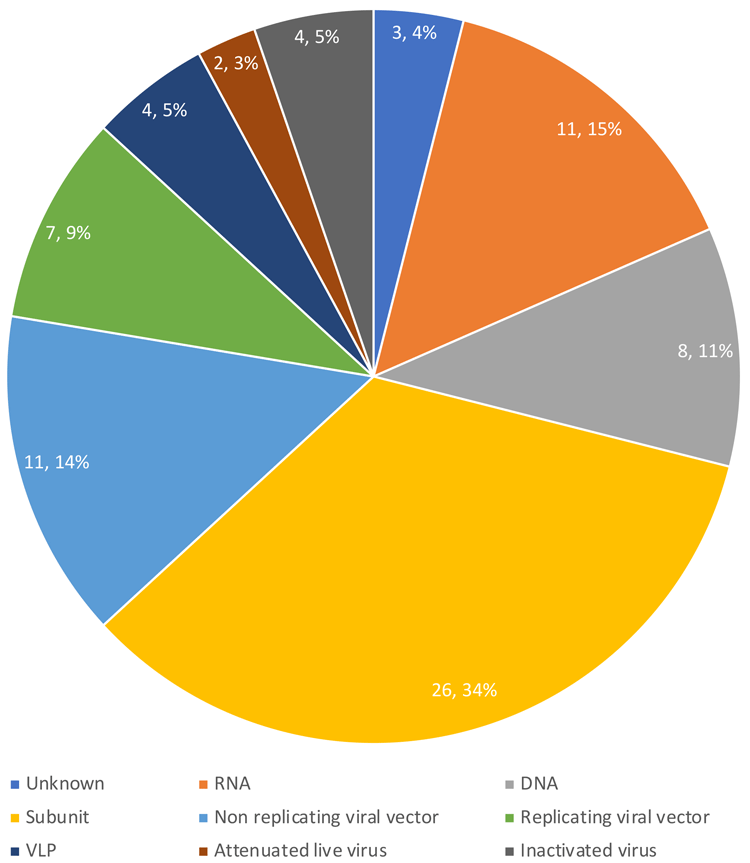
Figure 1. COVID-19 vaccines in development. Data from WHO, 20 April 2020
Specific examples of RNA vaccines in development include those by Moderna (Moderna–1273) and Imperial College (Professor R Shattock’s group). Examples of virally vectored vaccines include the Oxford/Jenner ChAdOx1 nCoV-19 and CanSino Biological adenoviruses. The reasons for focusing attention on these technologies for COVID-19 include their speed of development, scale and manufacturing fit as well as small dose size, all of which are highly desirable features for a rapid pandemic response.
Doses for different RNA vaccine formats vary. For self-amplifying mRNA (saRNA), dose levels of ca 1.25μg have been found effective in vaccines for influenza,1 while other mRNA formats may require doses in the range of 50-250μg.2 Virally vectored vaccines, where DNA for the antigen is delivered to the patient using a non-pathogenic viral vector (ie, adenovirus), typically require levels of 2.5 x 1010 viral particles per dose.3 Both RNA and virally vectored vaccine approaches have benefited from investments in cell and gene therapies over the last 10-20 years; in comparison, typical dose levels for more traditional vaccines range from 30-50μg/dose.4 For contract neutralising antibodies, which are meant to block cell receptors or viral antigens, higher dosing is typically required at around 10‑15mg/kg, equating to approximately 100mg for a person weighing 70kg.
To understand how the generation of antigens in a patient’s own cell has the potential to change vaccine production, a comparison of approaches is made in the next section.
Vaccine technologies
RNA vaccine technologies are attractive for the quick generation of a vaccine in response to rapid pathogen emergenceAs our scientific understanding has increased, vaccine technologies have advanced from classical live or attenuated vaccines to those dependant on recombinant platforms, such as subunit and virus-like particle (VLP) vaccines. Tools developed around cell and gene therapy applications have benefitted from technology convergence, enabling the delivery of genetic information (DNA and RNA) to produce antigens in cells, instead of replacement therapeutic proteins. This fundamentally differs from approaches used previously, whereby the antigen is delivered in its final form (higher therapeutic dose). Instead, the genetic information is supplied directly to the patient’s own cells, enabling them to effectively manufacture their own vaccine (lower therapeutic dose).
RNA vaccine technologies are attractive for the quick generation of a vaccine in response to rapid pathogen emergence
There are seven basic technology variants for producing vaccines and Table 1 illustrates the complexities of process selection. Each vaccine type has different development complexities, timelines and manufacturing requirements. Additionally, intrinsic safety profiles can differ among them.
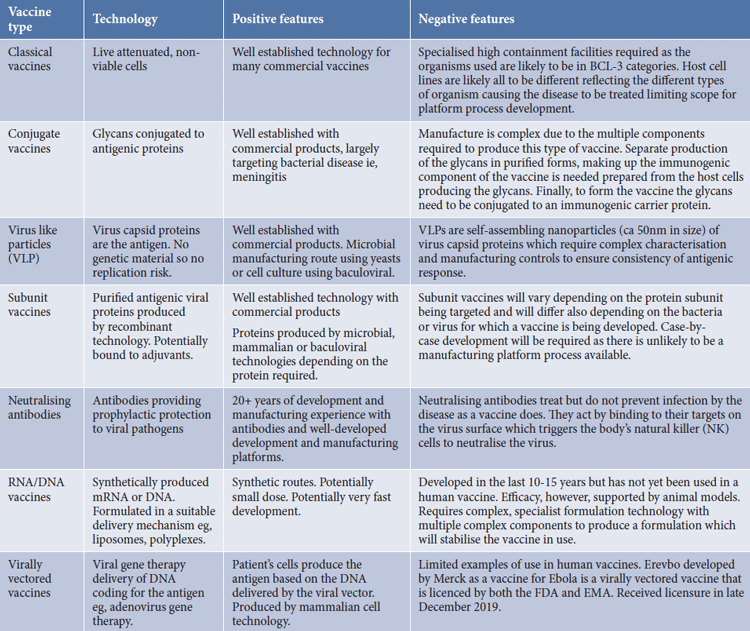
Table 1. Summary of vaccine technologies and features
Viral vector delivery of DNA
Viral vector vaccines use a replication-deficient virus to deliver a transgene encoding the antigen of interest for expression in the recipient’s cells. In principle, the manufacture of such vectors should be similar, regardless of the encoded antigen. Among the major classes of viral vectors, adenoviruses are arguably the most versatile and potent in inducing a combined antibody and T-cell response. Adenoviruses are large, non-enveloped viruses that can contain large transgenes with a total molecular weight of approximately 15MD per particle. They have an icosahedral protein capsid that encompasses a linear duplex DNA genome of approximately 36,000 base pairs.
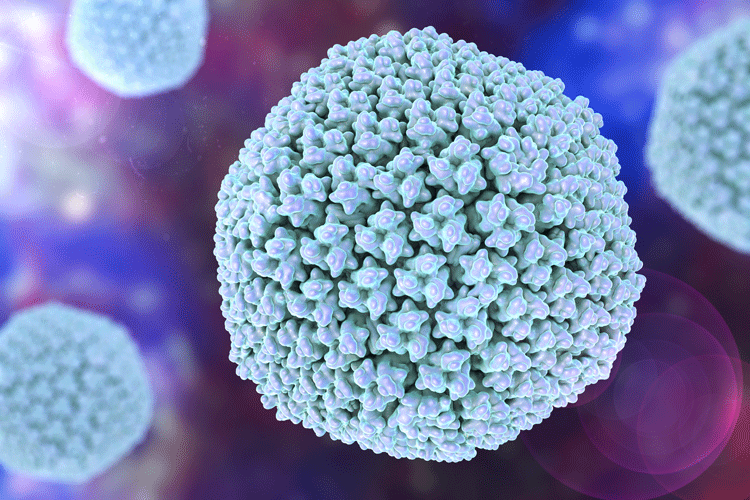
Figure 2. Model of an adenovirus
There are approximately 12 capsid proteins and no lipid envelope. The X-ray structure of the virus has been determined5 and a model is shown in Figure 2. Viruses lacking genes in the E1 and E3 regions are replication-incompetent outside helper cells, which provides a high level of assurance against the generation of replication-competent virus particles.
The growth of gene therapy has been supported by significant investment from contract development and manufacturing organisations (CDMOs) and the in-house capacity of large biopharmaceutical companies, together with the development of high-yielding and costeffective large-scale adenovirus production processes. The first virally vectored vaccine was licensed in the US and EU in 2019 against Ebola (Erevbo). The Oxford ChAdOx1 nCoV-19 vaccine, which is currently in clinical trials, builds on the experience gained during the development of adenovirus-based gene therapy vaccines, including oncolytic vaccines, plus extensive experience of good manufacturing practice (GMP)-compliant adenovirus production and manufacture.
There are marked differences between the growth characteristics of different viruses with varying adenovirus serotypes and antigen transgenes, which necessitates optimisation of production processes for each individual virus vector. This is clearly undesirable for low-cost manufacture or rapid outbreak response.
mRNA synthesis is highly productive, yielding in excess of 2g/L of full-length mRNA under optimal conditions
Human embryonic kidney 293 (HEK293) cells are frequently used in the production of viral vectors and vaccines. Suspension HEK293 cells are commonly used in batch-based stirredtank upstream processes with perfusion-based processes in development. Equipment for upstream processing is similar to that required for other mammalian cell culture systems, such as antibody production by Chinese hamster ovary (CHO) cells. Following growth of the HEK cells, the virus is introduced into the bioreactor resulting in cell infection and viral replication. Downstream processing then follows particular steps developed for gene therapy, which involves cell harvest, crossflow filtration and two different chromatography functionalities. Recombinant adenovirus serotype 5 (AdV5) is one of the most studied adenovirus vectors; however, to minimise issues with population immunity to human adenoviruses, the ChAdOx1 nCoV-19 vaccine is based on a chimpanzee adenovirus. This process is outlined in Figure 3.
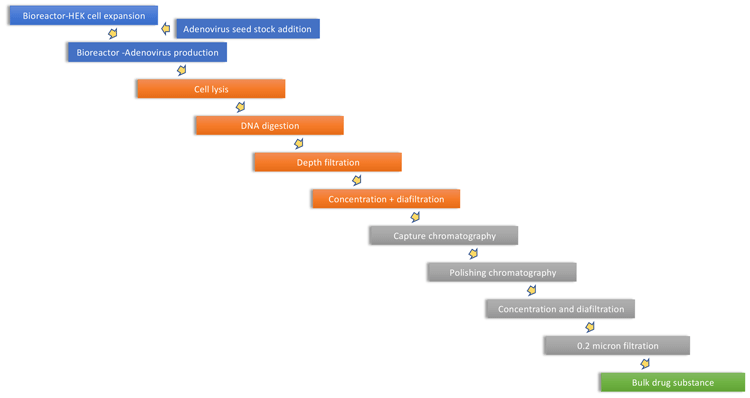
Figure 3. Adenovirus manufacturing process
Indications of production requirements for an adenovirus vaccine for COVID-19 can be extrapolated using data from two references. The first3 provides an indication of dose levels in terms of total particles from adenovirus vectored ChAdOx1 nCoV-19 vaccine, which is being tested for prevention of COVID-19 in non-human primates (NHP). The dosing described for human tests is 5e10 virus particles. A second publication6 describes the platform process performance (USP and DSP) to produce the adenovirus. The bioreactor sizing required to produce 1m vaccine doses is approximately 1,250L. To put this into perspective, there have been multiple installations of 2,000L single-use bioreactors for monoclonal antibody (mAb) production along with facilities providing multiple bioreactors at scales of over 10,000L. This demonstrates there is significant industry experience of large-scale cell culture operations. Although mAbs are typically produced using CHO, the requirements for HEK cell growth are similar. Therefore, this underlying capacity could be readily adapted for the production of adenovirus vaccines. The ChAdOx1 nCoV-19 vaccine entered Phase l/ll trials in April 20207 and preparations for scaling up to produce millions of vaccine doses have been established, with agreements between AstraZeneca and Oxford Biomedica.8
RNA vaccines
Another approach for producing antigens in cells involves using RNA instead of DNA. RNA vaccine technologies are attractive for the quick generation of a vaccine in response to rapid pathogen emergence and they are considered safe as they are unable to integrate into the host genome. They also have a short half-live in vivo, meaning that in vivo production of the antigen is finite and does not continue for long periods of time, reducing the impact on cellular function. Unlike DNA vaccines, which must cross two membranes to reach the nucleus to function, RNA vaccines only have to cross a single membrane to reach the cell cytoplasm for functionality. A review of mRNA vaccines has been published by Pardi.9
saRNA technology has the potential for wide application beyond in vivo production of antigenic proteins such as the in situ production of therapeutic proteins
mRNA vaccines are also produced by cellfree synthesis from a DNA template; therefore, the need for complex and time-consuming cell-line development is unnecessary. The ratio of mRNA generated per DNA template by in vitro transcription reactions is high (>1000x).10 Therefore, the level of plasmid DNA required to generate a DNA template for the production of a given amount of mRNA is small. This increases transfection efficiency and should subsequently have a knock-on effect on immunogenicity. RNA vaccines further combine features of safety with fast, totally cell-free production; furthermore, they induce both B- and T-cell responses. Two main mRNA vaccine platforms are currently available: synthetic mRNA molecules encoding only the antigen of interest and self-amplifying RNA (saRNA). saRNA utilises virally derived elements and encodes both the antigen of interest and viral proteins, enabling mRNA replication.11
An mRNA vaccine that has received a lot of attention is Moderna-1273, produced by Moderna Pharmaceuticals, which reached its first inhuman trials by late March with the following phases being accelerated.12 Preparations for large-scale production of the vaccine have been announced in collaboration with Lonza Biologics.13 The saRNA approach is being pioneered with the vaccine developed by Imperial College London (Shattock group) and is scheduled to be in Phase I trials in mid-June 2020. Scale up of the manufacturing process for large-scale dose production is being carried out by CPI (Centre for Process Innovation) in the UK.
Both platforms have been shown to induce an immune response, but it is not clear which approach is optimal. In current studies, both platforms are protective, but equivalent levels of protection were achieved using 1.25mg saRNA1 based on data from vaccines tested in animal models for other viral diseases compared to 50‑250μg for mRNA2 for COVID-19. The potential to achieve immunity using very small doses is a key reason for interest in saRNA as a platform for vaccines against viral diseases.
Using a cellular-like mRNA encoding the protein of interest provides immediate translation of the antigen but, although adequate antiviral protection has been shown, a large dose of synthetic mRNA material is required. To overcome this, several approaches have been developed based on a deeper understanding of cell biology. RNA consists of four nucleotides – guanine, uracil, adenosine and cytosine – and is designed to have a short half-life in vivo, acting as a carrier of information between DNA and proteins. One challenge of using RNA as a therapeutic has been controlling this half-life to guarantee therapeutic efficacy. Several methods have been developed to achieve this and here we focus on two methods: nucleotide modification and the use of saRNA. In addition to the RNA configuration, which alters dose levels, all RNA formats require advanced formulation technology to ensure efficacy by protecting the mRNA from RNase activity and facilitating RNA passage across cell membranes into the cell.
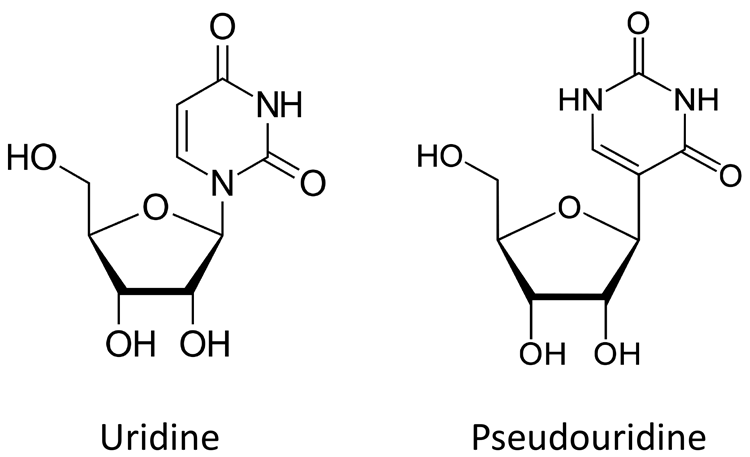
Figure 4. Structure of uridine and pseudouridine
Nucleotide modification – a demonstrated modification that has been found to increase biological stability of mRNA is the substitution of uridine by pseudo uridine.14 Comparison of the structure of uridine and pseudouridine is shown in Figure 4. This is a modification that can occur in vivo and increases the half-life of the mRNA. In vitro transcription used to generate mRNA easily achieves this substitution; pseudouridine is added to the transcription reaction, rather than uridine. This technology is believed to be the basis of the mRNA vaccines being developed by Moderna Biopharmaceuticals.
saRNA – an alternative strategy to mitigate the short half-life of mRNA is increased in situ mRNA production using saRNA.11 This makes use of elements from alphaviruses, which are positive‑strand, non-segmented RNA viruses. The alphaviral genome is divided into two open reading frames (ORFs): the first encodes proteins for the RNA-dependent RNA polymerase (replicase) and the second encodes structural proteins. In saRNA vaccine constructs, the ORF encoding viral structural proteins is replaced with any antigen of choice, while the viral replicase remains as an integral part of the vaccine and drives intracellular amplification of the RNA after immunisation. This is shown diagrammatically in Figure 5, which also displays a non-amplifying mRNA structure for comparison. In addition to demonstrating the two basic approaches for an mRNA vaccine, Figure 5 also denotes the preferred formulation of mRNA vaccines – lipid nanoparticles (LNPs).
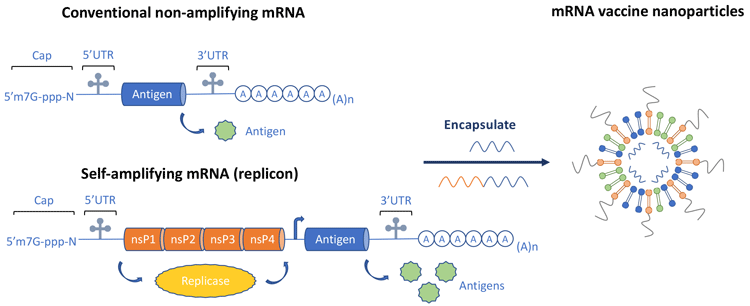
Figure 5. Comparison of conventional non amplifying mRNA and self-amplifying mRNA
LNPs have been found to be the most effective mRNA formulation approach;15 they protect the mRNA from degradation when injected into the patient and promote entry of the mRNA into cells. LNPs typically consist of four components: an ionisable cationic lipid, which promotes selfassembly into virus-sized (~100nm) particles and enables endosomal release of mRNA to the cytoplasm; lipid-linked polyethylene glycol (PEG), which increases the half-life of formulations; cholesterol, a stabilising agent; and naturally occurring phospholipids, which support the lipid bilayer structure. LNPs are potent tools for in vivo delivery of saRNA and conventional, non-replicating mRNA.
LNPs are potent tools for in vivo delivery of saRNA and conventional, non-replicating mRNA
mRNA production is facilitated by enzymatic in vitro transcription (IVT), which uses a DNA template such as a plasmid. mRNA production is therefore an enzymic reaction highly decoupled from biological expression, which is used for production of plasmid DNA and the enzymes used during in vitro synthesis. Depending on the specific mRNA construct and chemistry, the protocol may be adapted to use modified nucleosides, capping strategies or DNA template removal. To initiate the manufacturing process, template plasmid DNA produced in Escherichia coli is linearised using a restriction enzyme to allow synthesis of transcripts with a poly A tail attached at the 3′ end, increasing mRNA stability. Following template linearisation, the mRNA is synthesised from nucleotide triphosphates (NTPs) by a DNA-dependent bacteriophage RNA polymerase (such as T7, SP6 or T3). Once transcription is complete, the template DNA is digested by incubation with DNase. To enable efficient translation in vivo, the mRNA produced must be enzymatically or chemically capped. This capping can be either conducted postmRNA synthesis or more efficiently as part of the synthesis reaction.
mRNA synthesis is highly productive, yielding in excess of 2g/L of full-length mRNA under optimal conditions.16 Once the mRNA is synthesised, it is processed through several purification steps to remove reaction components including enzymes, free nucleotides, residual DNA and truncated RNA fragments. Production of mRNA starting from a plasmid template is shown schematically in Figure 6.
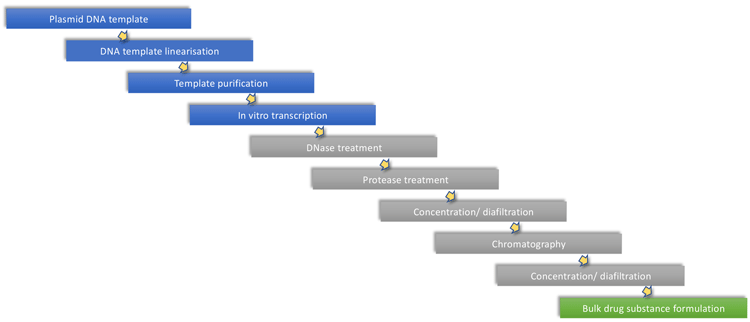
Figure 6. General mRNA production process outline
The attraction of mRNA, and particularly saRNA, relates to the small dose level (therefore minimising manufacturing scale), lack of dependence on biological systems (strains, cell lines) for production and the scope for platform process development (mRNA length being the main difference between different products). saRNA technology has the potential for wide application beyond in vivo production of antigenic proteins such as the in situ production of therapeutic proteins. An example of this is work evaluating saRNA to produce therapeutic antibodies in vivo.17
The small scale required to manufacture an saRNA vaccine can be estimated using available literature data. Based on a reaction yield of 2g/L,10 a dose level of 1.25μg1 and assuming a 50 percent purification yield, an IVT reaction volume of ca 1.3L would be sufficient to produce 1m vaccine doses. The small scale of the process, with the advantages of being easy to site a manufacturing process, procedure compactness and the lack of large manufacturing infrastructure, indicates why this approach to vaccine production is so attractive.
Conclusions
The promise of virally vectored DNA and mRNA vaccines, which use a patient’s own cells to produce antigens to pathogenic organisms, is tremendous in terms of vaccine safety and speed of response to new pathogens. The technologies used are the same as approaches being developed for cell and gene therapy, differing only in the length of time the transgene is required to be active in the patient’s cells. For a vaccine to be successful, this only needs to be long enough to raise antigens in the patient. For gene therapy applications, however, long-term expression of the transgene is desirable. This convergence of different therapeutic areas suggests they should not be considered as separate entities and that patients would benefit from pooling learning gained from production, delivery and therapeutic responses.
About the author
 John M Liddell is Chief Technologist at the Centre for Process Innovation (CPI), a UK-based technology innovation centre and part of the UK Catapult network. He has experience of a wide range of biopharmaceuticals, including novel protein formats, antibodies, viral vectors and vaccines. He provides support to biopharmaceutical development to increase access to new therapies through the application of innovative technologies.
John M Liddell is Chief Technologist at the Centre for Process Innovation (CPI), a UK-based technology innovation centre and part of the UK Catapult network. He has experience of a wide range of biopharmaceuticals, including novel protein formats, antibodies, viral vectors and vaccines. He provides support to biopharmaceutical development to increase access to new therapies through the application of innovative technologies.
John’s previous experience involved the development and manufacturing of biologics, working in senior process development roles with global organisations, including Fujifilm Diosynth Biotechnologies, Merck &Co, Avecia Biologics, Zeneca and ICI. He has almost 30 years of experience in bioprocessing with direct involvement in all phases of bioprocess development from early phase process definition to late phase process validation and commercial production.
References
1. Vogel et al “Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses.” Mol Ther. 2018 Feb 7;26(2):446-455
2. Evaluate Pharma May 19, 2020 “Moderna reminds the markets of biotech’s dual purpose”
3. van Doremalen et al ChAdOx1 nCoV-19 vaccination prevents SARS-CoV-2 1 pneumonia in rhesus macaques bioRxiv preprint doi: https://doi. org/10.1101/2020.05.13.093195
4. Branton et al “Teaming up for Vaccines” The Chemical Engineer May 2020 Issue 947
5. Reddy et al “Crystal Structure of Human Adenovirus at 3.5 A Resolution” Science. 2010 Aug 27;329(5995):1071-5.
6. Douglas et al “Simian Adenovirus Vector Production for Early-Phase Clinical Trials: A Simple Method Applicable to Multiple Serotypes and Using Entirely Disposable Product-Contact Components” Vaccine. 2019 Nov 8;37(47):6951-6961.
7. ClinicalTrials.gov A Study of a Candidate COVID-19 Vaccine (COV001)
8. Oxford Biomedica Signs Clinical & Commercial Supply Agreement with AstraZeneca, for manufacture of COVID-19 vaccine candidate, Oxford Biomedica press release, 28th May 2020.
9. Pardi et al “mRNA Vaccines – A New Era in Vaccinology” Nat Rev Drug Discov. 2018 Apr;17(4):261-279.
10. New England Biolabs application note, “RNA Synthesis: Tools to Take You from Template to Transcript”
11. Ljungberg K, Liljeström P “Self-replicating alphavirus RNA vaccine”, Expert Rev Vaccines. 2015 Feb;14(2):177-194
12. ClinicalTrials.gov Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-CoV-2 Infection (COVID-19).
13. Evaluate Pharma May 01, 2020 “Lonza to do the heavy lifting for Moderna’s Covid-19 vaccine”
14. Karikó et al “Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability”. Mol Ther. 2008 Nov;16(11):1833-1840.
15. Kowalski et al “Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery” Mol Ther. 2019 Apr 10;27(4):710-728.
16. Kigawa et al “A Multiphysics Model of in Vitro Transcription Coupling Enzymatic Reaction and Precipitation Formation” Biophys J. 2012 Jan 18;102(2):221-30.
17. Schlake et al “mRNA: A Novel Avenue to Antibody Therapy?” Mol Ther. 2019 Apr 10;27(4):773-784
Issue
Related topics
Biopharmaceuticals, Bioprocessing, Clinical Development, Clinical Trials, Drug Development, mRNA, Research & Development (R&D), RNA, Vaccines, Viruses






Hi Brian;
Thanks for raising excellent points, and I would appreciate it if you may please share the answer to your excellent questions.
Best regards
Mohammed Jawad
I have concerns about rna and its “half-life”. How long do the rna strands linger for in the cells? For how long do they express proteins? Are there health risks to excess proteins over time?