Improving quality in investigator‑led clinical trials
Posted: 26 August 2020 | Laura Trotta (CluePoints) | No comments yet
Laura Trotta explains why risk-based quality management is the best strategy to ensure data integrity of information from investigator-led clinical trials.

Introduction
Evidence generated by clinical trials, particularly randomised controlled trials (RCTs), is vital to inform medical practice. Considered the gold-standard approach for evaluating therapeutic interventions, RCTs allow for inferences about causal links between treatment and outcomes.1 Some trials are initiated by industry sponsors, such as pharmaceutical companies or clinical research organisations (CROs), whereas others, investigator-led trials, originate within research sites. The difference between the two usually centres on purpose. Industry-sponsored trials investigate experimental drugs with largely unknown effects and typically have an explanatory approach. They tend to be suitable for the development of novel agents or combinations. Whereas, investigator-led studies are more pragmatic; they usually investigate the benefits and harms of treatments in routine clinical practice. Table 1 characterises some of the contrasts between an explanatory and a pragmatic approach to clinical trials.
Table 1: Explanatory versus pragmatic approach to clinical trials2
Approach | Explanatory | Pragmatic |
Type of trial | Industry-sponsored | Investigator-led |
Primary purpose of trial | Regulatory approval | Public health impact |
Patient selection | Fittest patients | All patients |
Effect of interest | “Ideal” treatment effect | Actual treatment effect |
Endpoint ascertainment | Centrally reviewed | Per local investigator |
Preferred control group | Untreated (when feasible) | Current standard of care |
Experimental conditions | Strictly controlled | Clinical routine |
Volume of data collected | Large, for supportive analyses | Key data only |
Data quality control | Extensive and on-site | Limited and central only |
Industry-sponsored trials are usually highly efficient and benefit from pharma’s highly organised structures, both in terms of safety and regulatory compliance. Yet commercial interests and market expectations mean they may not always address all patients’ needs. Strictness of the eligibility criteria, the choice of comparators, effect size of interest and outcomes, as well as insufficient data on long-term toxicity, can all be restrictive.3 Arguably, the general principles underlying marketing approval by regulatory agencies, such as the Japanese Pharmaceutical and Medical Devices Agency (PMDA), the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) contribute to these limitations.
Shining a light on investigator‑led clinical trials
 Whether a new drug is sufficiently safe and effective for clinical use needs careful review. It requires meticulous assessment of the quality of the pivotal trial design, conduct, data and analysis. However, additional post-approval evidence on novel drugs or devices is still needed to provide a full understanding of the effectiveness and safety of competing interventions in ‘real life’. Investigator-led clinical trials allow for the assessment of patients and settings not necessarily covered by the initial approval, leading to potential extensions of indications and refinement of the drug usage in patient subgroups. Even for newly approved drugs, many questions of clinical interest typically remain unanswered at the time of approval, including the duration of therapy, dose or schedule modifications that may lead to a better benefit/risk ratio, combinations of the new drug with existing regimens, etc.
Whether a new drug is sufficiently safe and effective for clinical use needs careful review. It requires meticulous assessment of the quality of the pivotal trial design, conduct, data and analysis. However, additional post-approval evidence on novel drugs or devices is still needed to provide a full understanding of the effectiveness and safety of competing interventions in ‘real life’. Investigator-led clinical trials allow for the assessment of patients and settings not necessarily covered by the initial approval, leading to potential extensions of indications and refinement of the drug usage in patient subgroups. Even for newly approved drugs, many questions of clinical interest typically remain unanswered at the time of approval, including the duration of therapy, dose or schedule modifications that may lead to a better benefit/risk ratio, combinations of the new drug with existing regimens, etc.
Equally, repurposing of existing drugs, whose safety and efficacy profile is well documented in other indications, is often less complicated in investigator-led trials when compared to pharmaceutical companies that might have a product that ceases to be financially attractive towards the end of its life-cycle. In addition, large, simple trials that address questions of major public health importance have been advocated for decades as one of the pillars of evidence-based medicine.4
Crucially, more and larger investigator-led trials are required and it is essential to identify ways of conducting them as cost-effectively as possible. Investigator-led clinical trials have particular merit in the field of oncology, offering the potential to generate much of the evidence upon which the treatment of cancer patients is decided. Yet investigator-led trials may be under threat because of excessive regulation and bureaucracy and the accompanying costs. In addition, the contrasts between industry-led and investigator-led trials have direct implications on their conduct, notably with regards to ways of ensuring their quality.
Spiralling costs
…investigator-led trials should collect radically simpler data than industry-sponsored trials”
Clinical trial costs are rising. Recent estimates suggest that pivotal clinical trials leading to FDA approval have a median cost of $19 million; such costs are even higher in oncology and cardiovascular medicine, as well as in trials with a long-term clinical outcome, such as survival.3 In industry-sponsored trials, vast resources are spent in making sure that the data collected are error free. This is typically done via on-site monitoring (site visits) including source-data verification (SDV) and other types of quality assurance procedures, together with centralised monitoring including data management and statistical monitoring. While some on-site activities make intuitive sense, their cost has become exorbitant in the large multicentre trials that are typically required for the approval of new therapies. It has been estimated that for large, global clinical trials, leaving aside site payments, the cost of on-site monitoring represents around 60 percent of the total trial.4
The solution: RBQM
If monitoring activities significantly impacted patient safety or results, then high costs could be justified. Yet there is little evidence showing that extensive, intensive data monitoring has any major impact on the quality of clinical trial data. The most time-consuming and least efficient activity is source data verification (SDV), which can take up to 50 percent of the time spent for on-site visits. The monitoring of clinical trials needs to be re-engineered, not just for investigator-led trials, but also for industry-sponsored trials. To instigate and support this much-needed transition, regulatory agencies worldwide have advocated the use of risk-based quality management (RBQM), including risk-based monitoring (RBM) and central statistical monitoring (CSM).5,6
The central principle of RBQM is to “focus on things that matter”. What matters for a randomised clinical trial is to provide a reliable estimate of the difference in efficacy and tolerance between the treatments being compared. Crucially the criteria to assess efficacy and tolerance may differ between industry-sponsored trials and investigator-led trials.
 In terms of safety, investigator-led trials can collect much simpler data than industry-sponsored trials of drugs for which safety has not yet been demonstrated. Typically, in investigator-led trials, the occurrence of Common Terminology Criteria for Adverse Events grade three or grade four toxicities will suffice, plus any unexpected toxicity not known to be associated with the drug being investigated. Finally, medical history and concomitant medications, which may be important to document drug interactions with an experimental treatment, serve no useful purpose in investigator-led trials. All in all, investigator-led trials should collect radically simpler data than industry-sponsored trials.
In terms of safety, investigator-led trials can collect much simpler data than industry-sponsored trials of drugs for which safety has not yet been demonstrated. Typically, in investigator-led trials, the occurrence of Common Terminology Criteria for Adverse Events grade three or grade four toxicities will suffice, plus any unexpected toxicity not known to be associated with the drug being investigated. Finally, medical history and concomitant medications, which may be important to document drug interactions with an experimental treatment, serve no useful purpose in investigator-led trials. All in all, investigator-led trials should collect radically simpler data than industry-sponsored trials.
Equally, data quality needs to be evaluated in a “fit for purpose” manner. While it may be required to attempt to reach 100 percent accuracy in all the data collected for a pivotal trial of an experimental treatment, such a high bar is by no means required for investigator-led trials, as long as no systematic bias is at play to create data differences between the randomised treatment groups (for instance, a higher proportion of missing data in one group than in the other). Both types of trials may benefit from CSM of the data:
- industry-sponsored trials to target centres that are detected as having potential data quality issues, which may require an on-site audit
- investigator-led trials as the primary method for checking data quality.
Understanding CSM
CSM is a vital part of RBQM. As shown in Figure 1, the process starts with a Risk Assessment and Categorisation Tool (RACT). CSM helps quality management by providing statistical indicators of quality based on data collected in the trial from all sources. A “Data Quality Assessment” of multicentre trials can be based on the simple statistical idea that data should be broadly comparable across all centres. This idea is grounded in the fact that data consistency is an acceptable surrogate for data quality. Other additional tools of central monitoring can be used to uncover situations in which data issues occur in most (or sometimes all) centres, such as “Key Risk Indicators” and “Quality Tolerance Limits”. Taken together, all these tools produce statistical signals that may reveal issues in specific centres.
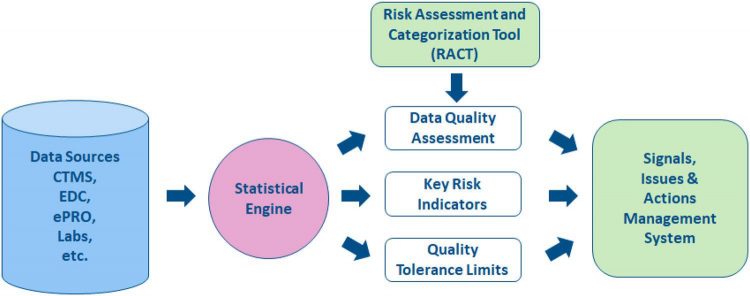
Figure 1: The Risk-Based Quality Management process
Actions must then be taken to address these issues, such as contacting the centre for clarification or in some cases performing an on-site audit to understand the cause of the data issue. Although it is a simple idea to perform a central data quality assessment based on the consistency of data across all centres, the statistical models required to implement the idea are necessarily complex to properly account for the natural variability in the data. Essentially, a central data quality assessment is efficient if:
- Data have undergone basic management checks, whether automated or manual, to eliminate obvious errors (such as out-of-range or impossible values) that can be detected and corrected without a statistical approach
- Data quality issues are limited to a few centres, while the other centres have data of good quality
- All data are used, rather than a few key data items such as those for the primary endpoint or major safety variables
- Many statistical tests are performed, rather than just a few obvious ones such as a shift in mean or a difference in variability.
The last two points are worth highlighting. It is statistically preferable to run many tests on all data collected than on a few data items carefully selected for their relevance or importance. Volume rather than clinical relevance is crucial for a reliable statistical assessment of data quality. The power of statistical detection comes from an accumulation of evidence, which would not be available if only important items and standard tests were considered. In addition, investigators pay more attention to key data (such as the primary efficacy endpoint or important safety variables), which therefore do not constitute reliable indicators of overall data quality. Nevertheless, careful checks of key data are also essential, although such checks generally are not statistical in nature.
CSM in use
Experience from actual trials as well as extensive simulation studies has shown that a statistical data quality assessment is effective at detecting issues with study conduct and data reliability. Experience from actual trials suggests that such study issues can be broadly classified as:
- Fraud, such as fabricating patient records or even fabricating entire patients
- Data tampering, such as filling in missing data or propagating data from one visit to the next
- Sloppiness, such as not reporting some adverse events, making transcription errors, etc
- Miscalibration or other problems with automated equipment.
Whilst these data errors impact trial results in different ways, all of them can potentially be detected using CSM, at a far lower cost and with much higher effectiveness than through labour-intensive methods such as SDV and other on-site data reviews. Investigator-led trials generate more than half of all randomised evidence on new treatments and it seems essential that this evidence be submitted to statistical quality checks before going to print and influencing clinical practice.
Conclusion
Investigator-led clinical trials are pragmatic trials that aim to investigate the benefits and harms of treatments in routine clinical practice. These much-needed trials represent most trials currently conducted. They are however threatened by the rising costs of clinical research, which are in part due to extensive trial monitoring processes that focus on unimportant details. Conversely, RBQM focuses instead on “things that really matter”. CSM plays a crucial role in RBQM, helping to drive down the cost of randomised clinical trials, especially investigator-led trials, while simultaneously improving their quality.
About the author
Dr Laura Trotta joined CluePoints in 2015 and moved into her current role as R&D manager in 2018, where she leads a team of data scientists responsible for developing new statistical and machine learning algorithms to assess the quality, accuracy and integrity of clinical trial data. Laura holds a Master’s degree in Biomedical Engineering and a PhD in Applied Mathematics from the University of Liège, Belgium.
References
- Collins R, Bowman L, Landray M, et al (2020) The magic of randomization versus the myth of real-world evidence. N Engl J Med 382:674–678
- Buyse M, Trotta L, Saad E, at al (2020) Central statistical monitoring of investigator‑led clinical trials in Int. J. Clin. Oncol 25:1207–1214
- Naci H, Davis C, Savovic J et al (2019) Design characteristics, risk of bias, and reporting of randomised controlled trials supporting approvals of cancer drugs by European Medicines Agency, 2014–16: cross sectional analysis. BMJ 366:5221
- Yusuf S, Collins R, Peto R (1984) Why do we need some large, simple randomized trials? Stat Med 3:409–422
- Moore TJ, Zhang H, Anderson G, et al (2018) Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015–2016. JAMA Intern Med 178:1451–1457
- Institute of Medicine (US) (2010) Forum on drug discovery development and translation. Transforming Clinical Research in the United States. National Academies Press, Washington DC
- European Medicines Agency (2011) Reflection paper on risk based quality management in clinical trials. Eur Med 1:94–103
- S. Department of Health and Human Services (2013) Food and drug administration guidance for industry. Oversight of Clinical Investigations. A Risk-Based Approach to Monitoring. National Academies Press, DC