Science and risk-based specification setting for excipients
Posted: 27 October 2020 | Bastiaan Dickhoff (DFE Pharma), Kalyan Janjanam (Epic Pharma), Sunil Kumar Nataraj (DFE Pharma) | No comments yet
Reducing unnecessary regulatory burden relating to excipients is important for any pharmaceutical manufacturer. In this article, Sunil Kumar Nataraj, Bastiaan Dickhoff and Kalyan Janjanam discuss a rational basis for controlling and specifying conflicting excipient attributes to avoid potentially tedious and complex regulatory processes.

Abstract
The COVID-19 pandemic has caused significant disruptions to current global supply chain systems and forced many companies to rethink their high reliance on China. It is of paramount importance to secure supply chain security and this should be addressed in the drug product application; however, there is often inappropriate specification of the excipients and/or inadequate science and risk-based justification for these properties. Where the supplier of an excipient was specified in an approved application, changing to a new supplier is an annual reportable but only “if the excipient specification remains unchanged”. It is therefore critical to appropriately specify the excipient in the initial application for regulatory flexibility. Exercising control via specification does not absolve the applicant from the responsibility of having an appropriate science and risk-based change control, as the impact from switching from one excipient source to another may depend on attributes that could not have been previously specified.
Pharmacopoeial compliance is a minimum safety standard, so it is not usually an issue when sourcing excipients from multiple sources. However, pharmacopoeial monographs may reference other attributes, such as particle size, without specifying limits. These occur under the non-mandatory functionality related characteristics section of European Pharmacopoeia (Ph Eur) monographs or the additional requirements (labelling) section of the monographs in the United States National Formulary (USP-NF). Suppliers provide their own methods and limits for such attributes, as well as other attributes not referenced by the pharmacopoeia. It is tempting to adopt the specification from the first qualified supplier of an excipient, but this may prove restrictive on qualifying future suppliers, who may specify different methods and limits for the non-mandatory attributes.
Considering recent publications – ICH Q12 and KASA – a rational basis to specify excipients using multivariate oversight will be discussed below.
Introduction
ICH Q6A1 describes specification as “a list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described”.
Specifications are critical quality standards that are proposed and justified by the applicant and approved by regulatory authorities. For any change, applicants must assess the effects of the change on product quality through appropriate studies.2 ICH Q123,4 guidance (Figure 1) addresses the gaps in technical and regulatory aspects that limit a flexible regulatory approach to post-approval chemistry, manufacturing and controls (CMC) changes. Although the reporting mechanism for many CMC changes is clear, the US Food and Drug Administration (FDA) is concerned that there is confusion regarding which elements of an application are considered to be established conditions (ECs).3,4 This confusion could have a negative impact on change management activities and discourage continual improvement of product manufacturing processes, leading to unnecessary submission of post-approval supplements to the FDA for changes that could be managed by existing market authorisation holder’s (MAH’s) quality systems.
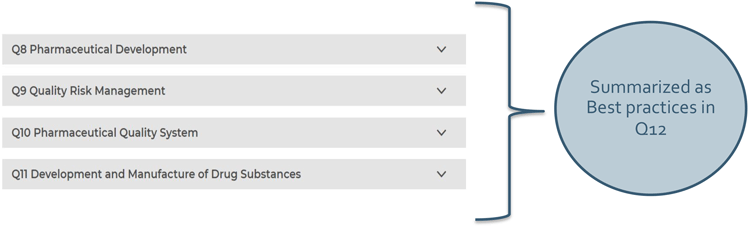
Figure 1: Q12 encompasses best practices of ICH Q8 through Q11
A common misconception is that pharmacopoeial methods must be used to demonstrate compliance; the use of alternative methods, which are suitably validated, is in fact permitted by USP-NF and Ph Eur. ECs are described under appendix 1 of ICH Q12 (Figure 2 and 3). Acceptance criteria for some critical material attributes (CMAs), contributing to ECs, may be a part of labelling requirements.

Figure 2: Q12 distinguishes between ECs and those which may not be considered as ECs under the control strategy
It is important to note that all regulatory submissions contain a combination of ECs and ‘supportive information’. An MAH is expected to proactively make an explicit identification and submission of combinations of ECs and specify appropriately any change of category level and related justification in the original submission or consequent annual reports. The decision tree mentioned in Q12 could help identify ECs with associated reporting categories. However, issues arise with implicit ECs, where being incorrectly identified in earlier communications and any proposed change by MAHs in post-approval communications to the regulatory agency potentially causes additional regulatory burden.

Figure 3: Excerpt from the appendix 1 of ICH Q12 describing what constitutes an EC and what does not under the control strategy for excipients
Discussion
ECs are legally binding and considered necessary to assure product quality. Q12 necessitates that the below topics are captured:
- Summary of product control strategy.
- Reporting category for making changes to approved ECs. The reporting category should be based on MAH’s justification or available guidances.
- Post approval change management protocol (PACMPs) at the time of submission or prospective submission, eg, having a Multivariate (MV) oversight for post-approval CMC commitments.
The FDA, in pursuit of constant improvement, proposed a new knowledge-aided assessment and structured application (KASA)5 initiative (Figure 4). This guidance may be used by applicants to devise a structured data presentation considering not only question-based review (QbR) and quality by design (QbD), but also have correctly identified ECs, eg, specification setting of an immediate release (IR) excipient. Control strategies should provide additional insights (level of detail) into the design of experiments/ design space and risk assessments, inclusive of chemometrics monitoring, eg, using MV analyses.
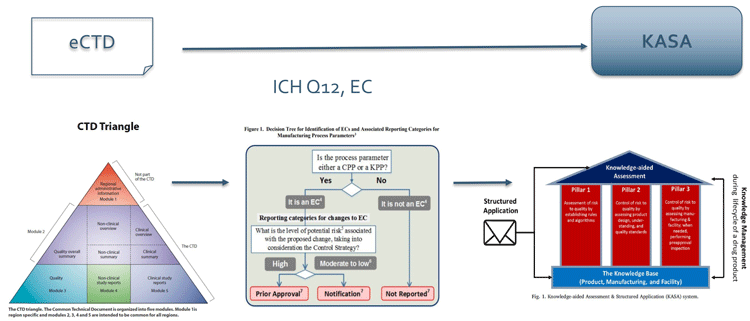
Figure 4: Regulatory flexibility with KASA: ICH Q12 is seen as intermediary preparatory step towards KASA from current electronic Common Technical Dossier (eCTD)6-based drug submissions
KASA is expected to facilitate drug approvals and post-approval changes for both regulators and applicants. It is also expected to streamline the process and facilitate applicants/MAHs, from product launch and throughout the product lifecycle, backed by correctly identified ECs in the first submission and/or subsequent revisions.
Some salient features of KASA are:
- Assure patient-focused quality standards with science- and risk-based regulatory approaches through established algorithms
- Use and compare computer-aided analyses to regulatory standards and evaluate quality risks against approved applications/facilities. Automatically retrieve historical data and facility information, thus providing better evaluations and decisions, eg MV analyses.
- Reduce the subjectivity of documentation and time liabilities; therefore, improving the quality and efficiency of assessments
- Improve the objectivity of regulatory actions and provide more consistent communication
- Identifying risks, mitigation and control strategy during product lifecycle management (PLM).
Conclusion
MAHs conducting alternate supplier development programmes must look at science- and risk‑based finished product quality when comparing IR excipients from one supplier versus another. This would ensure ECs are met with appropriate justification during post-approval change, revisions or annual reports. As part of the application review process, the FDA is expected to assess the proposed EC in conjunction with the level of product detail and process understanding, including the MAH’s risk assessment activities. Demonstration of risk mitigation within the application could allow for greater operational and regulatory flexibility with correctly identified and listed ECs.
These measures could enable “excipient portability” for a change within the same grade of excipient eg, a spray dried excipient from one supplier to another meeting the same finished product quality based on correctly identifying ECs in the original submission or acquired knowledge post-approval using the comparability protocol.7
Furthermore, for listed ECs, MAHs would be encouraged to set specifications for a product’s functionality (eg, consistent multiyear product trends for dissolution) rather than a paper-based exercise (matching individual IR excipient attributes, eg, apparent PSD etc), which are usually part of labelling requirements of USP-NF. It is important that an MAH is in control of a specification for incoming IR excipients and therefore should define acceptance criteria, preferably based on the intended finished product quality. Adopting an excipient supplier’s specification will result in having to make any post-approval changes and associated correspondence with the regulatory agency for approvals – which is a tedious and complex process.
KASA is expected to facilitate drug approvals and post-approval changes for both regulators and applicants”
It should be noted that certain product-specific drug applications may need thorough investigation, eg, a sustained release (SR) formulation based on a certain hydroxy propyl methyl cellulose (HPMC) grade of one excipient manufacturer versus a post-approval change of the same SR formulation employing a different HPMC grade from another supplier. Such changes may warrant the comparability protocol to be approved by the regulatory agency before the MAH can continue manufacturing the finished product with requested changes. In contrast, for IR formulations using an excipient within the same grade, post-approval changes documented per comparability protocol, with established equivalency/product sameness considering any variability in particle size of IR excipients, are known to cause little regulatory issues, as demonstrated by Kushner et al.8
For legacy products for which the MAH did not submit an original application with a clear delineation of the EC, the FDA intends to develop a process by which applicants could obtain clarification regarding ECs. This is important, given the fact that no two excipient suppliers manufacture an excipient employing the same equipment, analytical test methods or identical QC release specifications, including that of labelling requirements. An MAH should consider this and ensure that an alternate supplier development programme is in place if they are largely dependent on a single excipient supplier.
MV analyses-based product development has been around for many years and Figure 5 demonstrates a robust tabletting by wet granulation process with unit operations monitored and controlled by MV modelling.9
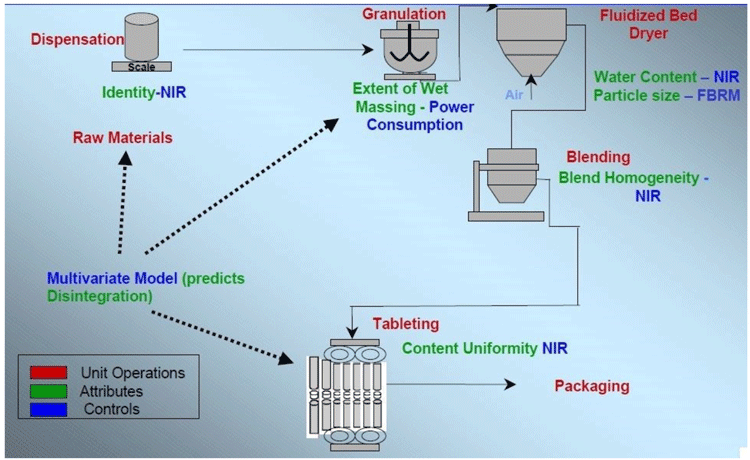
Figure 5: Demonstration of a robust tabletting by wet granulation process with unit operations monitored and controlled by MV modelling (Fritz Erni, Novartis)9
For establishing design space (R&D) and verification of design space (scale up) for specified attributes through to having long-term commercial success (manufacturing) for specified and unspecified attributes with MV analyses (Figure 6), applicants/MAHs largely rely on excipient manufacturer’s process capability and excipient supplier’s advice on application related CMAs. This ensures product robustness throughout the product lifecycle, which is further augmented by studying multiyear10 trend data (MV analyses).
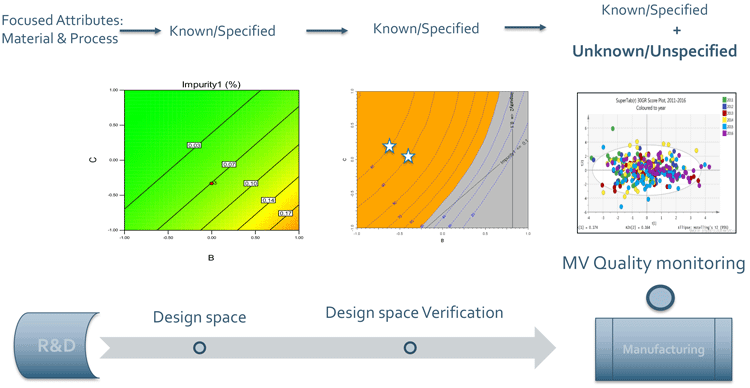
Figure 6: MV analyses helps to identify, monitor and control specified and unspecified attributes. ICH Q12 and the KASA initiative recommends MAHs have an MV oversight for post‑approval changes.
In summary, smart specification for excipients can be achieved with correctly identified CMAs and by setting acceptance criteria (inclusive of updated knowledge gained post approval via annual reports) which constitutes ECs. Having an MV oversight together with implementing elements of KASA can help applicants/MAHs mitigate excipient variability risks, especially for IR formulations, and reduce post‑approval regulatory burden.
About the author
Sunil Kumar Nataraj is a pharmaceutical leader with on-the ground experience of providing problem-solving solutions across the US, Europe, Japan and India. Sunil joined the Technology & Innovation department at DFE Pharma in 2017 as product application specialist in Germany. Sunil has pharmaceutical product development expertise in oral solids focusing on ODT’s and continuous twin screw granulation supporting continuous manufacturing. Sunil focuses on enhanced customer R&D interaction catering to current Pharma market trends and changing Pharma regulatory landscapes.
Co-authors: Bastiaan Dickhoff and Kalyan Janjanam.
References
1. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/ Biological Products Q6b. Mar 1999.
2. Guidance for Industry Process Validation: General Principles and Practices. Current Good Manufacturing Practices (CGMP),Revision 1, January 2011.
3. Technical and regulatory considerations for pharmaceutical product lifecycle management, ICH Q12 Nov 2019
4. Established Conditions: Reportable CMC Changes for Approved Drug and Biologic Products. FDA. May 2015 Pharmaceutical Quality/CMC.
5. Knowledge-aided Assessment & Structured Application (KASA): A New Approach that Modernizes FDA’s Quality Assessment of Regulatory Drug Applications. FDA. Sept 2018.
6. The common technical document for the registration of pharmaceuticals for human use: quality – M4Q(R1) quality overall summary of module 2 modules 3: quality. Sept 2002.
7. Comparability Protocols —Chemistry, Manufacturing, and Controls Information. FDA, Feb 2003
8. Quality-by-Design Study for an Immediate-Release Tablet Platform: Examining the Relative Impact of Active Pharmaceutical Ingredient Properties, Processing Methods, and Excipient Variability on Drug Product Quality Attributes. Kushner at al. Journal Of Pharmaceutical Sciences 103:527–538, 2014
9. Design space and control strategy by Fritz Erni, (Novartis Basel). Presented at SAQ, Olten 2006
10. Multivariate analysis in the pharmaceutical industry: enabling functional excipient parameters data into knowledge. Sunil Kumar N, DFE Pharma, AAPS 2018.