

Maintaining EU MDR certification: a spotlight on post‑market clinical follow-up
Posted: 2 November 2021 | Celeste Maksim (RQM+) | No comments yet
Celeste Maksim, Chief of Staff, Clinical and Post-Market Practice, RQM+ advises on strategising for PMCF compliance, highlighting areas to prioritise to ensure an efficient process.

THOUGH THE deadline for compliance with the EU Medical Device Regulation 2017/745 (MDR) has passed, manufacturers must maintain processes and documentation to preserve their CE marking. The regulation places greater emphasis on post‑market clinical follow-up (PMCF) – an area that requires ongoing attention. Manufacturers were able to opt for an extended deadline of May 2024; however, they are not exempt from PMCF compliance in the meantime. All devices must comply with EU MDR post-market requirements even if they have a renewed certificate under the previous medical device directive (the criteria to benefit from the 2024 deadline).
Why is PMCF so important?
PMCF activities enable manufacturers to confirm the safety and performance of a device even once it is being used by patients or healthcare professionals, and to do so throughout its expected lifetime. Through PMCF, manufacturers can ensure the device achieves its intended use and clinical benefit, monitor side effects that they are already aware of, but also identify any new risks, thereby ensuring that the device’s benefit-risk ratio remains acceptable. Moreover, real-world use reveals whether devices are being used properly by end users, according to the instructions provided, so that manufacturers can identify misuse or off-label use. Pharmaceutical businesses must therefore fully understand PMCF requirements, particularly if they:
- Also manufacture medical devices, such as devices with ancillary medicinal substances
- Partner with or supply to companies that manufacture medical devices
- Manufacture a drug that is sold pre-packaged in a delivery device.
This article provides advice on planning for PMCF compliance and highlights important considerations along the way.
Planning for PMCF success
 Since there are higher expectations for the quality and quantity of data under the EU MDR, considerable resources may be required to implement PMCF requirements. The key to delivering optimal results is good planning, since adequate preparation will ensure that there is no sacrifice of quality. It is highly recommended that manufacturers begin by planning a clear strategy from clinical, post-market/quality, business and operational perspectives that can be used to compile a PMCF plan; this document must be included within the post-market surveillance plan. According to EU guidance,1 ‘a PMCF plan shall specify the methods and procedures to proactively collect and evaluate clinical data from the use in or on humans of a CE-marked medical device.’
Since there are higher expectations for the quality and quantity of data under the EU MDR, considerable resources may be required to implement PMCF requirements. The key to delivering optimal results is good planning, since adequate preparation will ensure that there is no sacrifice of quality. It is highly recommended that manufacturers begin by planning a clear strategy from clinical, post-market/quality, business and operational perspectives that can be used to compile a PMCF plan; this document must be included within the post-market surveillance plan. According to EU guidance,1 ‘a PMCF plan shall specify the methods and procedures to proactively collect and evaluate clinical data from the use in or on humans of a CE-marked medical device.’
Building a strategy
Manufacturers’ PMCF strategy should be clearly documented and include: an activity timeline, available budget, the risk class of each device, the quality of data needed to provide sufficient evidence, and whether a general or specific PMCF activity is required to provide the data needed. It should be noted that most devices will require both specific and generic PMCF activities. The activities must be selected according to the residual risks identified through clinical evaluation, and there must be sound justification for these selections, as well as measurable objectives. In summary, manufacturers must aim to build the strongest possible, minutely documented, rationale for PMCF data inclusion or exclusion, depth and relevance, which can be easily discussed and defended with notified bodies. In cases where time does not permit the ‘ideal’ PMCF activity, it is recommended that manufacturers propose a multi‑year approach to demonstrate how the required quality data will be obtained through a series of planned PMCF activities. 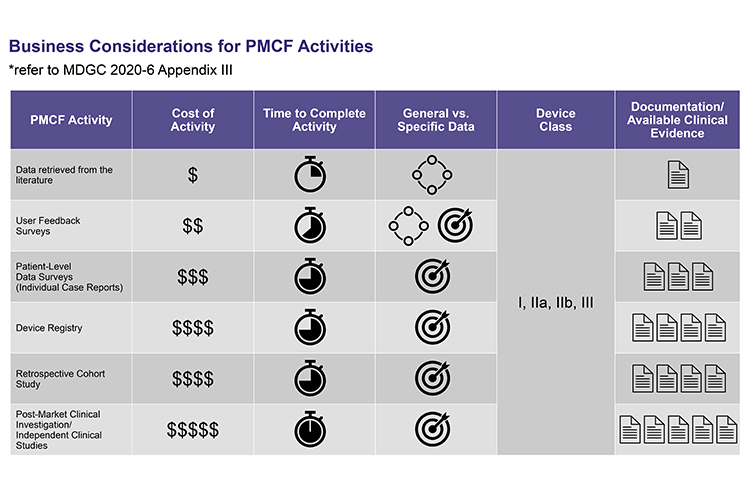
Commercial considerations
It is advisable to thoroughly assess business considerations for each PMCF activity from the outset:
- Timeline for PMCF activity
- Timeline to update supporting documents based on requirements for the risk classification of the device (PMS Report/PSUR, CER, SSCP [if applicable])
- Budget
- Resource needs.
Regulatory professionals should assess the ideal combination of people, systems, support and skills needed, and then work from there. It may be more efficient to outsource some activities, either to allow internal resources to focus on daily operations or to avoid training staff on specialist skills that are only temporarily needed. They must, however, bear in mind that skills and resources may be in short supply, with manufacturers working towards deadlines for the MDR and EU In Vitro Diagnostic Devices Regulation (IVDR), and this will affect planning.
Prioritising activities
 The planning process can also be used to prioritise devices according to the certificate expiration timeline, gap analysis, likely lifecycle, and the product’s role within larger therapeutic systems. Resources can then be allocated accordingly, taking into consideration the volume of devices requiring data remediation, as this may affect how responsibilities are divided within product teams. It may not be commercially viable to pursue MDR compliance for some product classes, at least in the short term, in which case it is best to make this decision as early as possible and then establish a proactive plan for compliance. For example, if high quality clinical data on a product is needed but is not available for the device, then the PMCF activity in Year 1 may be to set up a registry or system to obtain access to the data or to understand what type of data is available and if a specific activity format is feasible (retrospective patient‑chart review). Then the PMCF activity in Year 2 could be to collect the data, expand or pursue a different option based on the outcome of Year 1. Ultimately, attempting to achieve compliance without proper planning or sufficient resources could result in the business having to withdraw products from the market under pressure.
The planning process can also be used to prioritise devices according to the certificate expiration timeline, gap analysis, likely lifecycle, and the product’s role within larger therapeutic systems. Resources can then be allocated accordingly, taking into consideration the volume of devices requiring data remediation, as this may affect how responsibilities are divided within product teams. It may not be commercially viable to pursue MDR compliance for some product classes, at least in the short term, in which case it is best to make this decision as early as possible and then establish a proactive plan for compliance. For example, if high quality clinical data on a product is needed but is not available for the device, then the PMCF activity in Year 1 may be to set up a registry or system to obtain access to the data or to understand what type of data is available and if a specific activity format is feasible (retrospective patient‑chart review). Then the PMCF activity in Year 2 could be to collect the data, expand or pursue a different option based on the outcome of Year 1. Ultimately, attempting to achieve compliance without proper planning or sufficient resources could result in the business having to withdraw products from the market under pressure.
Consistent processes
The next step in the preparatory stage is to establish standardised processes for PMCF data assessment. It will be easier to organise activities across different teams if processes and the way information is presented remains consistent. Clinical data should be organised to clarify what data is available for each device, medical indication and target population. It may be necessary to update the clinical evaluation first, to identify relevant data. Once achieved, standardisation will also facilitate feeding information into other areas of post-market monitoring, such as complaint trends, adverse events or field safety corrective actions. The benefit of a uniform process is that the clinical story of the product will be consistent across all documents in the submission and those supporting ongoing compliance.
Data assessment
 Once PMCF activities are under way and the required data has been sourced, manufacturers will need to identify residual or emerging risks, and then assess where additional clinical evidence is needed. They must then identify the most effective method for gathering this evidence, ensuring that the method is appropriate and will address the risks identified. It can be a challenge for manufacturers to determine whether they have sufficient clinical evidence, as the amount of clinical evidence required will largely depend on the risk class of the device, the indication, claims, available data to support the device, and any recent changes in clinical practice or the device itself. When reviewing the results of PMCF activities, manufacturers should also pay close attention to quality and relevance, ensuring that the data collected or literature cited is meaningful, supports device safety, performance and intended use, and is likely to meet notified body expectations for the risk class of each device.
Once PMCF activities are under way and the required data has been sourced, manufacturers will need to identify residual or emerging risks, and then assess where additional clinical evidence is needed. They must then identify the most effective method for gathering this evidence, ensuring that the method is appropriate and will address the risks identified. It can be a challenge for manufacturers to determine whether they have sufficient clinical evidence, as the amount of clinical evidence required will largely depend on the risk class of the device, the indication, claims, available data to support the device, and any recent changes in clinical practice or the device itself. When reviewing the results of PMCF activities, manufacturers should also pay close attention to quality and relevance, ensuring that the data collected or literature cited is meaningful, supports device safety, performance and intended use, and is likely to meet notified body expectations for the risk class of each device.
Efficient review
It is worth noting that, although notified bodies cannot legally offer consultancy, it is possible to clarify the intent of their questions, or the meaning of nonconformities raised. However, careful and thorough planning as outlined above, as well as sound decision-making and documentation, helps to reduce notified body review times and queries. According to notified body feedback, a common pitfall among manufacturers is failing to include enough detail in their documentation, which drives up the number of questions during review. The rationale for selecting a PMCF activity needs to be included, despite the reasons seeming obvious. The documentation submitted should show precisely how much data is available for each device by device variant, indication and population. The documentation should explain why the level and type of clinical data is appropriate, and also provide references to the EU MDR or relevant guidance to support the rationale. Since notified body numbers and capacity are severely limited, taking the time to provide this level of detail will favour an efficient review, saving time and resources for all parties.
About the author
Celeste Maksim has a PhD in analytical chemistry, RAC certification and over a decade of experience in regulated industries including pharmaceuticals, medical devices and in vitro diagnostic products. Celeste’s main focus at RQM+ is on building and managing PMS & PMCF/PMPF services.
Reference
- Medical Device Coordination Group Document, MDCG 2020-7, Post-market clinical follow-up (PMCF) Plan Template: A guide for manufacturers and notified bodies, April 2020.
Further information
For more helpful guidance on staying on track for EU MDR compliance, download the dedicated PMCF whitepaper from RQM+ here: https://hubs.ly/H0VJm5q0
Issue
Related topics
Industry Insight, Medical Devices, Regulation & Legislation, Technology





