

Applications of solid-state NMR spectroscopy to pharmaceuticals
Posted: 21 February 2013 | | No comments yet
Solid-state NMR spectroscopy (SSNMR) has emerged as an extremely powerful analytical technique for the characterisation of pharmaceuticals[1-5]. Despite its capability, SSNMR is still not used ubiquitously in the pharmaceutical industry. Several factors contribute to this, including cost and analysis time, but two of the major reasons are understanding the full capability of the technique and having access to the expertise necessary to acquire and interpret the data. In this article, the use of SSNMR to study small molecule pharmaceuticals and its application in preformulation, formulation and manufacturing stages of pharmaceutical development are studied. The basics of solid-state NMR spectroscopy will be described. In particular, the information content of a SSNMR spectrum will be highlighted, such as the chemical shift, line width and relaxation time. Specific nuclei will also be discussed, including the advantages of 13C, 19F, 1H and other less abundant nuclei. Finally, four applications will be highlighted, including polymorph identification, quantitation, amorphous characterisation and dynamics.
Why SSNMR?
In solid-state, the primary motivation is to determine those properties unique to solid drug substances and drug formulations. In particular, the questions that are often asked include:
- Is the material crystalline or amorphous?
- What is the structure of the crystalline form?
- Is there more than one crystalline form?
- How much of the different forms are present?
- What is the mobility of the material, especially in the amorphous state?
- Can these questions be answered for both the drug substance and drug product?
Solid-state NMR spectroscopy is uniquely suited to answer these questions. It is a non-destructive and non-invasive technique that is both quantitative and selective. It can be used to look at structure as well as dynamics. Each of these areas will be highlighted in greater detail below.
Non-destructive and non-invasive
While solid-state NMR spectroscopy can be used to look at all drugs, one of the greatest applications used for looking at drug formulated products and the advantage of solid-state NMR spectroscopy is that, for 13C NMR, the drugs usually have a different chemical shift from those of the excipients. Other nuclei, such as 19F and 31P, are not typically found in excipients and so only the drug is observed in the spectrum. In addition to drug formulations, SSNMR can be used to study drugs in inclusion compounds as well as drugs in polymer matrices, such as amorphous dispersions.
Quantitative and selective
SSNMR spectroscopy is inherently a quantitative technique[6]. This arises from the fact that the NMR signal is proportional to the number of nuclei that are present. This fact is often used in solution NMR spectroscopy to quantify methyl vs. methine vs. methylene functional groups. It is possible to quantify the different crystalline forms, including polymorphs and solvates, as well as mixtures of crystalline and amorphous forms. The advantage of SSNMR compared to most other analytical techniques is that standard curve is not needed in order to obtain quantitative data. Another advantage of SSNMR spectroscopy is the ability to selectively label compounds. For example, 13C is only 1.1 per cent natural abundance. Placing a 13C label in the molecule that is present at 100 per cent means that the signal from that particular nucleus will be about 90 times larger compared to the signals from the other 13C nuclei. This is useful for looking at drug excipient interactions, as well as changes upon formulation.
Structure
Solid-state NMR is particularly useful for looking at the structure of the compound. There is an emerging field of NMR crystallography in which the structure of the compound in the crystalline state can be determined based on various solid-state NMR experiments[2,4,7]. In addition, solid-state NMR can readily provide information about the number of crystallographically-inequivalent sites, as well as confirmation, hydrogen bonding and packing arrangement in the lattice.
Dynamics
Finally, solid-state NMR can provide information about the dynamics of the system. For crystalline systems, the relaxation times may provide information about the mobility of the molecules in the crystal lattice. For more office materials, the NMR relaxation time changes as the material approaches the glass transition temperature. The advantage of NMR spectroscopy is that mobility can be observed independently for the drug and the excipients, which is useful in the analysis of amorphous dispersions.
Basics of SSNMR
In NMR spectroscopy, the nuclei of atoms are irradiated with radio waves, and the corresponding resonance frequency of the nuclei is observed. The frequency of irradiation depends on the magnetic strength of an applied magnetic field and a constant called the magnetogyric ratio, which is different for each isotope. NMR spectrometers are often referred to by the resonance frequency for 1H NMR, with the highest field instruments having a frequency of 1000 MHz (or more). Higher field instruments can provide improved sensitivity and resolution, which is critical when dealing with mass-limited samples and/or high molecular weight proteins. In solution NMR, the line widths of the peaks are typically very narrow. This is primarily due to the rapid tumbling of the molecules in solution. In the solid state, however, the molecules are not tumbling rapidly, and two of the interactions that are averaged in solution, namely 1H-13C dipole-dipole couplings and chemical shift anisotropy (CSA), are no longer averaged. To compensate for this, high-power 1H decoupling is necessary to obtain high-resolution 13C NMR spectra in the solid state. In addition, there is a distribution of chemical shifts caused by the range of molecular orientations with respect to the static method, and this distribution can be up to 200 ppm broad. Spinning the sample at an angle of 54.7° with respect to the static magnetic field (called magic-angle spinning or MAS), averages the chemical shifts to their isotropic value. Finally, a technique called cross polarisa – tion (CP) is often used to enhance sensitivity and a shorter relaxation time.
Information content from SSNMR spectra
The primary piece of information available from the NMR spectrum is the chemical shift. The chemical shift is dependent on the local electronic environment of the molecule and is very sensitive to slight changes in molecular conformation, which is useful for identifying different crystalline forms. The NMR line width also provides useful information about the system. Amorphous line widths are typically about an order of magnitude broader than those of crystalline lines. Crystalline line widths can also provide information about the morphology of the system. The line shape can also be useful. For example, 13C lines will be asymmetric due to coupling to 14N. Relaxation times can provide information about dynamics of the system, especially as a function of temperature. In addition, relaxation times of crystalline materials have been correlated to particle size, and may have implications for predicting functional properties such as chemical stability and dissolution rate. Finally, two-dimensional NMR techniques are used to show connectivity between drugs and excipients as well as assigning peaks in the solid state, which is useful for determining miscibility of amorphous dispersions.
Practical aspects of SSNMR
As noted previously, sensitivity is often a critical component in the analysis of pharmaceuticals, especially for the detection of small amounts of one crystalline form either in the presence of another crystalline form or in the presence of amorphous material. Each nucleus has specific advantages and disadvantages when it comes to sensitivity. Sensitivity is dictated in part by the magnetogyric ratio, which determines the resonance frequency and scales almost quadradically with field strength. Although 1H NMR has the highest magnetogyric ratio of the commonly-observed nuclei, which is related to better sensitivity, and is almost 100 per cent abundance, resolution is a big problem, as obtaining a high-resolution spectrum can be difficult due to strong 1H-1H dipolar interactions. For this reason, 13C is the most commonly studied nucleus in the solid state. Both 19F and 31P NMR have the advantage of a high magneto – gyric ratio and high natural abundance, and because they are isolated in the molecule, will typically have narrow peaks. The downside is that they are not always present in the molecule.
When to perform SSNMR
The decision to perform solid-state NMR experiments depends upon several factors. Unlike some other analytical techniques, SSNMR experiments typically require tens to hundreds of milligrams of sample. For new drug substances, there may not be enough sample initially to perform solid-state NMR experiments. Another consideration when performing solid-state NMR experiments is the time and expense. Solid-state NMR experiments typically take hours to days to run, and therefore the throughput is much lower than other analytical techniques. The instrumentation is also about an order of magnitude more expensive than techniques such as differential scanning calorimetry (DSC).
Applications of SSNMR Spectroscopy
Four applications are highlighted to show some of the information that can be obtained from a SSNMR experiment. Further information about the capabilities of SSNMR can be found in several excellent review articles.
Polymorph identification
SSNMR is probably most recognised for its capability to identify different crystalline forms of a compound[1-5,7]. As noted previously, the chemical shift is very sensitive to slight changes in the local electronic environment and so different crystalline forms, whether they are polymorphs, hydrates or solvates, will have different SSNMR spectra. Figure 1 (below) shows 13C CPMAS NMR spectra of different crystalline forms of gabapentin. Forms II and III (A and B, respectively) are anhydrate forms of gabapentin, and there is sufficient resolution to identify each of the forms. Figure 1C shows a spectrum of a sample of Form II that had been ball milled, and contains approximately two per cent of Form III.
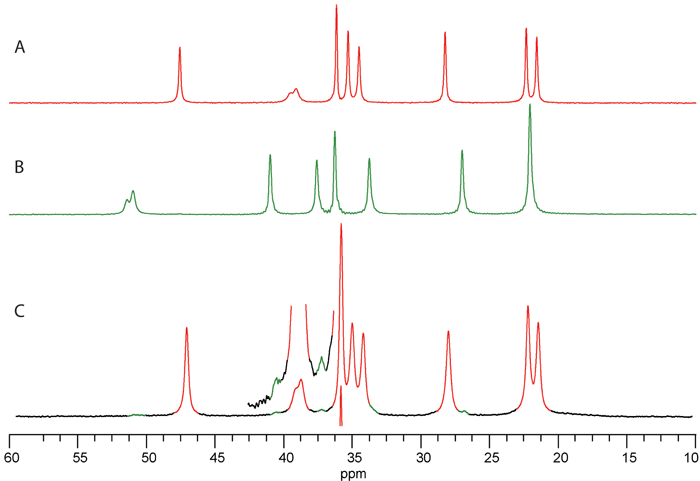
Figure 1. 13C CPMAS NMR spectra of different crystalline forms of gabapentin
Quantitation
There have been several examples highlighting the quantitative capability of SSNMR, with most of the work based upon 13C CPMAS NMR spectroscopy[6,8-13]. In an early example of the technique, Gao showed that delavirdine polymorphs and pseudopolymorphs could be accurately quantitated8. Wenslow highlighted the use of 19F MAS NMR to identify and quantify crystalline and amorphous forms in both bulk and in dosage forms.9 Offerdahl et al. showed that crystalline and amorphous forms of neotame could be quantified without the need for calibration curves[6]. More recently, Farrar et al. showed that crystalline material could be quantified in the presence of a liquid formulation[11]. Virtanen and Maunu showed that an active pharmaceutical ingredient could be detected at levels as low as 0.7 per cent[12]. Finally, Zielinska-Pisklak and co-workers showed that the amount of crystalline carvedilol Form II could be quantified in pharmaceutical dosage forms.[13]
Amorphous characterisation
The capability of SSNMR to study amorphous forms was demonstrated in several publica tions[10,14-20], and in particular, a recent study by Pham et al. highlighted the one and two-dimensional NMR capability to study amorphous solid dispersions18. Brus has also shown the capability of 19F NMR to characterise amorphous atorvastatin in dosage forms[19].
Dynamics
SSNMR has long been used to look at dynamics in amorphous systems, including lyophilised systems[17,20-23]. Yoshioka and co-workers showed the role of excipients in determining the glass transition temperature of lyophilised systems[21]. Others have focused on using relaxation times to probe molecular mobility and phase separation[15,17,18,23]. Figure 2 (below) shows a plot of relaxation time vs. 1/T for an amorphous dispersion of nifedipine and polyvinylpyrrolidone. The relaxation times of the drug and polymer start to decrease about 20° below the glass transition temperature, and continue to decrease, highlighting the capability of SSNMR to follow the mobility of the system around the glass transition temperature.
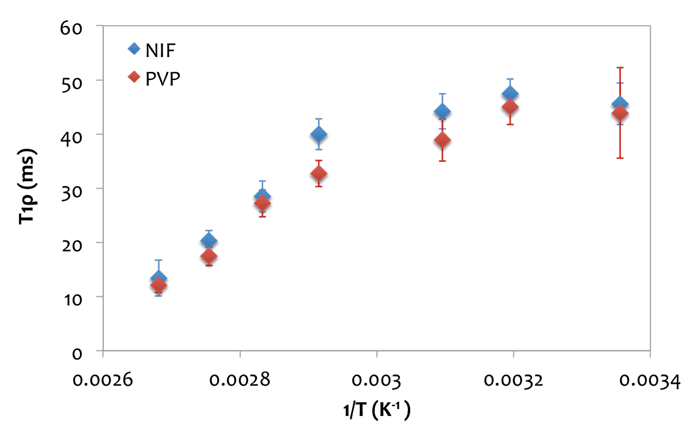
Figure 2: Relaxation time vs. 1/T for an amorphous dispersion of nifedipine and polyvinylpyrrolidone
Conclusion
SSNMR spectroscopy has many applications to the characterisation of pharmaceutical solids. Recent publications have highlighted the capability of SSNMR to identify polymorphs, provide quantitative data in both drug substance and drug product and study complicated formulations such as amorphous dispersions.
References
- Tishmack PA, Bugay DE, Byrn SR 2003. Solid-state nuclear magnetic resonance spectroscopy – Pharmaceutical applications. Journal of Pharma – ceutical Sciences 92(3):441-474
- Harris RK 2007. Applications of solid-state NMR to pharmaceutical polymorphism and related matters. Journal of Pharmacy and Pharmacology 59(2):225-239
- Geppi M, Mollica G, Borsacchi S, Veracini CA 2008. Solid-state NMR studies of pharmaceutical systems. Applied Spectroscopy Reviews 43(3):202-302
- Vogt FG 2010. Evolution of solid-state NMR in pharmaceutical analysis. Future Medicinal Chemistry 2(6):915-921
- Berendt RT, Sperger DM, Isbester PK, Munson EJ 2006. Solid-state NMR spectroscopy in pharmaceutical research and analysis. Trac-Trends in Analytical Chemistry 25(10):977-984
- Offerdahl TJ, Salsbury JS, Dong ZD, Grant DJW, Schroeder SA, Prakash I, Gorman EM, Barich DH, Munson EJ 2005. Quantitation of crystalline and amorphous forms of anhydrous neotame using C-13 CPMAS NMR spectroscopy. Journal of Pharmaceutical Sciences 94(12):2591-2605
- Harris RK 2006. NMR studies of organic polymorphs & solvates. Analyst 131(3):351-373
- Gao P 1996. Determination of the composition of delavirdine mesylate polymorph and pseudo – polymorph mixtures using C-13 CP/MAS NMR. Pharmaceutical Research 13(7):1095-1104
- Wenslow RM 2002. F-19 solid-state NMR spectroscopic investigation of crystalline and amorphous forms of a selective muscarinic M-3 receptor antagonist, in both bulk and pharmaceutical dosage form samples. Drug Development and Industrial Pharmacy 28(5):555-561
- Lefort R, De Gusseme A, Willart JF, Danede F, Descamps M 2004. Solid state NMR and DSC methods for quantifying the amorphous content in solid dosage forms: an application to ball-milling of trehalose. Int J Pharm 280(1-2):209-219
- Farrer BT, Peresypkin A, Wenslow RM 2007. Quantitation of crystalline material within a liquid vehicle using H-1/F-19 CP/MAS NMR. Journal of Pharmaceutical Sciences 96(2):264-267
- Virtanen T, Maunu SL 2010. Quantitation of a polymorphic mixture of an active pharmaceutical ingredient with solid state C-13 CPMAS NMR spectroscopy. Int J Pharm 394(1-2):18-25
- Zielinska-Pisklak M, Pisklak DM, Wawer I 2012. Application of C-13 CPMAS NMR for Qualitative and Quantitative Characterization of Carvedilol and its Commercial Formulations. Journal of Pharmaceutical Sciences 101(5):1763-1772
- Kimura K, Hirayama F, Arima H, Uekama K 1999. Solidstate C-13 nuclear magnetic resonance spectroscopic study on amorphous solid complexes of tolbutamide with 2-hydroxypropyl-alpha- and -beta-cyclodextrins. Pharmaceutical Research 16(11):1729-1734
- Forster A, Apperley D, Hempenstall J, Lancaster R, Rades T 2003. Investigation of the physical stability of amorphous drug and drug/polymer melts using variable temperature solid state NMR. Pharmazie 58(10):761-762
- Schachter DM, Xiong JC, Tirol GC 2004. Solid state NMR perspective of drug-polymer solid solutions: a model system based on poly(ethylene oxide). Int J Pharm 281(1-2):89-101
- Masuda K, Tabata S, Sakata Y, Hayase T, Yonemochi E, Terada K 2005. Comparison of molecular mobility in the glassy state between amorphous indomethacin and salicin based on spin-lattice relaxation times. Pharmaceutical Research 22(5):797-805
- Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS, Strohmeier M, Vogt FG 2010. Analysis of Amorphous Solid Dispersions Using 2D Solid-State NMR and H-1 T-1 Relaxation Measurements. Molecular Pharmaceutics 7(5):1667-1691
- Brus J, Urbanova M, Sedenkova I, Brusova H 2011. New perspectives of F-19 MAS NMR in the characterization of amorphous forms of atorvastatin in dosage formulations. Int J Pharm 409(1-2):62-74
- Luthra SA, Utz M, Gorman EM, Pikal MJ, Munson EJ, Lubach JW 2012. Carbon-deuterium rotational-echo double-resonance NMR spectroscopy of lyophilized aspartame formulations. Journal of Pharmaceutical Sciences 101(1):283-290
- Yoshioka S, Aso Y, Kojima S 1999. The effect of excipients on the molecular mobility of lyophilized formulations, as measured by glass transition temperature and NMR relaxation-based critical mobility temperature. Pharmaceutical Research 16(1):135-140
- Suihko EJ, Forbes RT, Apperley DC 2005. A solid-state NMR study of molecular mobility and phase separation in co-spray-dried protein-sugar particles. European Journal of Pharmaceutical Sciences 25(1):105-112
- Wojnarowska Z, Grzybowska K, Adrjanowicz K, Kaminski K, Paluch M, Hawelek L, Wrzalik R, Dulski M, Sawicki W, Mazgalski J, Tukalska A, Bieg T 2010. Study of the Amorphous Glibenclamide Drug: Analysis of the Molecular Dynamics of Quenched and Cryomilled Material. Molecular Pharmaceutics 7(5):1692-1707
Acknowledgement
I am a partial owner of Kansas Analytical Services, a company that provides solid-state NMR services to the pharmaceutical industry. The results presented here are from my academic work at the University of Kansas and the University of Kentucky, and no data from Kansas Analytical Services is presented here.
Biography
Eric Munson received his BA degree from Augustana College in Sioux Falls, South Dakota. After studying for a year in Munich, Germany, on a Fulbright Fellowship, he continued his education at Texas A&M University, where he received his PhD degree in 1993. He then spent one year as a postdoctoral fellow at the University of California, Berkeley, under the direction of Professor Alexander Pines. In 1994, he started as an Assistant Professor in the Chemistry Department at the University of Minnesota, and was promoted to Associate Professor in 2000. In 2001 he moved to the Pharmaceutical Chemistry Department at the University of Kansas, where he was promoted to Professor in 2006. He moved again in 2010 to the Department of Pharmaceutical Sciences at the University of Kentucky, where he is the Patrick DeLuca Endowed Professor in Pharmaceutical Technology. His research program is focused on the characterisation of pharmaceutical solids using a variety of analytical techniques, with an emphasis on solid-state NMR spectroscopy. He is a recipient of an NSF CAREER award and a McKnight Land-Grant Professorship, and is a Fellow of the AAPS.
