Gene to drugs: can expression be the key to new discoveries?
Posted: 25 February 2013 | | No comments yet
Cancer treatment faces a conundrum: a growing lack of therapeutics with lasting effects. The low hanging fruit of the medicinal chemistry orchard seems to have been picked, and modification of existing anti-cancer therapeutics has produced only incremental rewards[1]. Thus, both pharmaceutical companies and academic researchers are left searching for new strategies that will yield novel, long-lasting therapies and methods to rationally utilise existing therapies, alone and in combination, to individualise treatment regimens. The search for novel anti-cancer therapeutics or novel uses for existing drugs can benefit from the maturity of pharmacogenomics to extract genomic information for identification of new targets and therapeutic strategies.
Pharmacogenomic strategies interrogate the genome using low-throughput and high throughput platforms that generate quantifiable information from cells or tissues reflecting gene expression, protein expression, epigenetic changes or mutation / polymorphism status that correlate with therapeutic response. Herein, I will highlight the possibilities and challenges of utilising gene expression signatures as a tool to discover novel, anti-cancer agents and to optimise therapeutic regimens.
Gene expression information can be captured using high-density DNA microarrays and RNA sequencing (RNA-seq) technologies, each capable of nearly whole genome coverage. RNA-seq has an advantage over DNA micro arrays because this technology measures splice variants and provides unmatched base resolution that can link mutation status to gene expression levels[2]. With either platform, the scientist can amass catalogues of gene expression data from cell culture or animal models and human tumour samples. However, utilisation of gene expression levels requires sampling of target tissue, so most cancers are tractable for using gene expression analysis as a drug discovery and development tool (Figure 1).
Application of gene expression analysis in drug discovery: Finding novel targets
It is estimated that the human genome encodes several thousand ‘druggable’ genes. However, depending on the tumour / tissue type, the landscape of druggable genes may vary. Candidate therapeutic targets must be highly expressed and differentially-expressed in tumour tissue. Collection of gene expression data, followed by bioinformatic assessment of differentially-expressed genes, can initiate identification of new therapeutic targets followed by validation of potential candidates.
Expression signatures of messenger RNA (mRNA) and microRNA (miRNA) genes characterise normal cells or tissues from diseased counterparts. Sorting of the gene expression signatures that differentiate normal from diseased cells or tissues will identify mRNA, or protein-coding genes, highly expressed in diseased tissue. These represent candidates as novel targets for therapy. Using systems biology methods, candidates are sorted for genes expected to code for proteins having enzymatic activity or functioning as membrane-bound receptors or ion channels with the expectation that these proteins will be highly-expressed and accessible for pharmacological disruption.
Dysregulation of miRNA genes in diseased cells or tissue can also lead to new targets. miRNA hybridise to specific mRNA to control expression of the mRNA. Interrogation of target genes of miRNA genes over-expressed in diseased cells might indicate miRNA that are oncogenes or tumour suppressors. Oncogenic miRNA may be directly disrupted in tumour cells for therapeutic benefit.
A second approach to novel target discovery using the gene expression information from tumour cells or tissues exploits the complexity of the expression signatures, sometimes termed expression networks, rather than interrogating the expression levels on a gene-by-gene basis. Functional overlapping of biological networks is understood to allow cancer cells to thrive in the face of single gene / protein disruption. For example, non-small cell lung cancers with specific mutations in the epidermal growth factor receptor (EGFR) might initially respond to small molecule inhibitors of EGFR. Unfortunately, most of these patients progress due to the survival of tumour cells with compensatory mutations[3-5]. Deconstruction of expression signatures that characterise a disease phenotype, such as resistance to therapy, can be carried out using systems biology tools like DAVID and Ingenuity Pathway Analysis[6,7]. These tools help the experimentalist infer the biological function(s) of the signature genes and organise the signature genes into networks of activity. Like any network, from flight patterns of commercial aircraft to electrical systems, these expression networks contain ‘hub(s) of activity’. For drug discovery strategies, these hubs, whether signature genes or not, might represent novel drug targets.
‘Drug discovering’ strategies using expression data were benchmarked in bacterial and yeast systems to identify molecular interactions, and subsequently, targets of specific compounds. A nine-gene expression signature, derived from analysis of steady-state transcriptional levels of SOS repair pathway mutants, was used by Gardner and colleagues to identify mitomycin C targets in E. coli. The nine gene signature was compared with signatures that characterised mitomycin C-treated E. coli to uncover targets of drug activity[8]. Similarly, several research groups have used mRNA expression information to develop a catalogue of expression patterns corresponding to either mutation or pharmacological disruption of specific proteins in S. cerevisae with the goal of using gene expression data to understand the specificity of therapeutic compounds[9,10]. In practice, methods to overlay chemical and genetic signatures must first find matches and second, infer mechanism of action and potentially the target of the new compound. These experimental examples were forerunners of systematic approaches to identify the mechanism of action and target of novel agents based on gene expression signatures in humans.
Developing a catalogue of chemical genetic lesion gene expression signatures in prokaryotes and lower eukaryotes is relatively easy due to genome size and comparative absence of functionally-compensating genes. Identification of a chemical-genetic expression interaction catalogue in humans is more challenging because tissue-specific or cell-specific gene expression must be considered. Expression signatures characterising genetic lesions can be derived from cells treated with interfering RNA to elicit individual gene disruption or from diseased patient samples for which genetic mutation status is known. Steady state expression profiles are generated after compound treatment as described above and anchored to an experimental surrogate for a desired clinical phenotype, such as inhibition of cell proliferation. The resulting chemical-genetic expression profiles are overlaid.
The Golub group tested the chemical genetic interaction methodology on human cells by collecting gene expression data following chemical perturbation using high throughput screening (GE-HTS) of compounds with uncharacterised targets. Gene expression signatures of desired biological states of human cells were generated. For instance, a potential treatment for leukaemia was hypothesised as one that could induce differentiation of the leukemic cells. Gene expression signatures of differentiated blood cells were overlaid on expression signatures of leukaemia cells treated with a library of novel entities and monitored for differentiated phenotypes in vitro. This methodology provided data that linked activities of several compounds using the chemical-genetic interactions[11]. Similarly, Antipova and colleagues searched for inhibitors of platelet derived growth factor receptor (PDGFR) signalling in neuroblastoma cell lines. To mimic the desired phenotype of an inhibitor of PDGFR, cells were first treated with PDGF and subsequently treated with an inhibitor of MAPK signalling. A library of uncharacterised com – pounds was screened to identify entities that elicited a similar gene expression phenotype as an MAPK inhibitor, and two emerged with the desired properties[12].
The drug discovery efforts described above used a small set of signature genes for query. If a computationally manageable signature is not attainable or if complexity is desired to capture the biological system, how can the experimentalist manage these signatures to identify mechanism of action or targets of novel entities? As previously discussed, modelling of gene expression networks can lead to novel drug targets. Lamb and colleagues analysed gene expression data characterising individual genetic lesions, disease phenotypes, and response to small molecule insult with both supervised and unsupervised computational approaches to develop a ‘pattern matching’ algorithm[13]. The Connectivity Map (CMap) emerged and was used to generate a searchable database linking hundreds of expression profiles of small-molecule drug activities to outcome phenotypes. CMap was used to find inhibitors of androgen sensitive prostate cancers and identified small molecules that inhibit HSP90[14]. The Connectivity Map has also identified lead targets (HDAC, PI3K and others) for lung and breast cancer therapy that have been subsequently externally validated[15].
Gene expression signatures can also include non-coding RNAs. For example, miRNA expression signatures can indicate miRNA species that may be directly targeted or may function as novel therapeutic agents themselves. Signatures of miRNA expression that characterise drug treatment or a cellular state contain an additional layer of complexity because miRNA can target multiple mRNAs. Thus, multiple proteins may be affected by a single miRNA, depending on the cell or tissue type. Web-based algorithms (e.g. miRDB and TargetScan) can predict the probability that a specific miRNA will target specific mRNA and also demonstrate the spectrum of mRNAs targeted. The networks of cellular activity specified by an miRNA expression signature can also be utilised to identify hubs, either proteins or miRNAs, which might be novel targets of therapy.
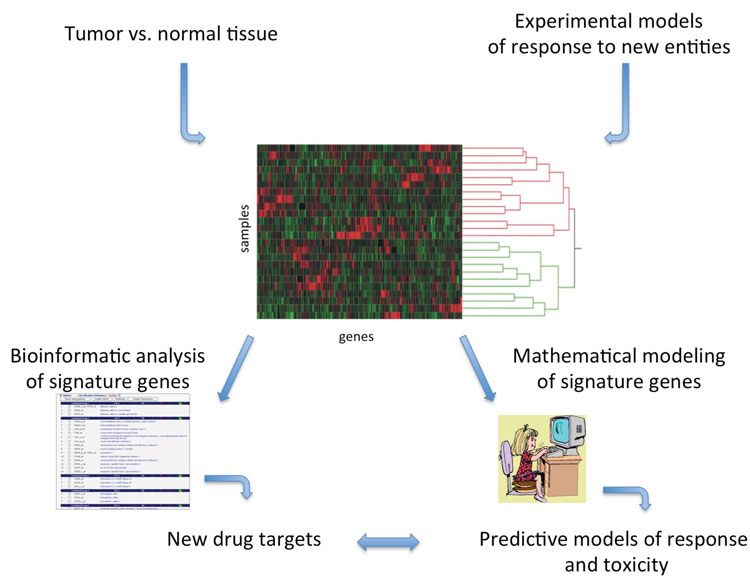
Figure 1: Drug discovery and development applications for differential gene expression signatures using experimental model systems or patient tumour samples. Expression signatures can be annotated to identify druggable targets or predictive models of response can be mathematically derived. Gene expression data can, in some cases, be utilised for both purposes from the same sample material
Identifying biomarkers of response and toxicity
Development of new drugs is one way of increasing the clinical stockpile of therapeutics, but rational and individualised use of existing drugs or drug combinations may also improve the clinical problem. I will explore the use of gene expression signatures as biomarkers of toxicity and response as a means for directing new and existing therapies through trials and into clinical use. Perhaps the real hurdle of drug discovery will be successful development of effective drug therapies that only show benefit in a minority of patients.
Developing new anti-cancer agents that demonstrate pre-clinical promise frequently derails in clinical trials for a host of reasons: 1) adverse events and toxicities that are too extensive to allow therapeutic success, such as cardiotoxicities, 2) low numbers of responders, in cases with EGFR inhibition in colorectal cancers, or 3) insignificant improvement over placebo or standard-of-care[16-19]. Individualisation of approved agent(s) and identification of rational combination(s) of anti-cancer agents can be achieved using gene expression information in conjunction with means for matching to activities of specific compounds, such as CMap. Strategies such as these may improve response to a drug by selecting responsive populations. As an example, our work and that of others has shown that identification of gene signatures that predict whether an individual is likely to respond to a given therapy can successfully stratify patients to an appropriate therapeutic regimen[20-22]. Using gene expression signatures identified in our lab that predict response to EGFR inhibition in lung cancers, we also asked whether the signature suggested additional signalling pathways that may be targeted in specific patients. Annotation of the signature for biological functions revealed that combination of EGFR and MEK inhibitors might benefit additional patients that individually demonstrated either low response or unacceptable toxicities to one of the drugs[20,23,24]. Thus, predictive signatures are also descriptive in that they may uncover activities that might be therapeutically targeted in combination (Figure 1 on page 10).
Finally, it should also be possible to develop gene expression signatures predictive of toxicities to anti-cancer agents. Existing pharmacogenetic biomarkers of toxicity, polymorphisms in drug metabolising enzymes that correlate with toxicity to specific therapies, have been clinically utilised with some success[25]. To develop a compendium of gene expression data from patient tumours that characterise toxicities, tumour sampling followed by gene expression analysis can and should be built into clinical trials. Signatures can be identified that characterise unacceptable toxicities in the same way as signatures of response. Thus, using a single platform for collecting gene expression data, signatures of toxicity and response can accompany drugs through later trials to quicken the pace of drug development.
Concluding remarks
Discovery of novel anti-cancer agents and development of individualised anti-cancer therapies are uniquely positioned to use gene expression data as a tool for identification of new drug targets and new uses for existing cancer therapies. Like most advances in science and medicine, risk is always part of the equation. Gene expression analysis requires sampling of diseased tissue, and tissue from liquid or solid tumours is often more readily available than normal tissue or from other disease states. However, collection of tumour tissue is not without some risk to the patient. The risk may be balanced by the valuable expression data acquired from these samples that may lead to new therapeutic regimens, new uses of old therapies, new targets for chemical entities, and new molecular markers for response and toxicity.
Lastly, new technologies, such as RNA-seq, not only allow for measurement of gene expression but also provide pharmacogenetic assessment of mutation status in coding regions of genes. In some cases, a single assay may then be used to merge multiple genomic inputs to develop new therapies and new biomarkers for a novel therapy. The end objective is to maximise the safety profile of drugs and individualise the therapeutic regimens to improve response profiles and limit adverse events in patients.
References
- D. Cressey. Traditional drug-discovery model ripe for reform. Nature. 2011. 471: 17-18
- Oshlack, A., Robinson, M.D., and M.D. Young. From RNA-seq reads to differential gene expression results. Genome Biol. 2010. 11(12): 220
- Kobayashi, S. et al., EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005. 352(8): 786-92
- Engelman, J.A., et al., MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007. 316(5827):1039-43
- Pao, W., et al., Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005. 2(3): e73
- Huang, D.W., Sherman, B.T., and R.A Lempicki. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009. 491):44-57
- Ingenuity® Systems, www.ingenuity.com
- Gardner, T.S., et al., Inferring genetic networks and identifying compound mode of action via expression profiling. Science. 2003. 301(5629):102-105
- Marton, M.J. et al., Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 1998. 4(11):1293-301
- Parsons, A.B., et al., Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004. 22(1):62-69
- Stegmaier, K., et al. Gene expression-based highthroughput screening (GE-HTS) and application to leukemia differentiation. Nat Genetics. 2004. 36(3): 257-263
- Antipova, A.A., Stockwell, B.R. and T.R. Golub. Gene expression-based screening for inhibitors of PDGFR signaling. Genome Biology. 2008. 9:R47
- Lamb, J., et al. The Connectivity Map: Using Gene-expression signatures to connect small molecules, genes, and disease. Science. 2006. 313(5795):1929-1935
- Hieronymus, H., et al. Gene expression signaturebased chemical genomics signature identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006. 10:321-330
- Qu, X.A. and D.K. Rajpal. Applications of Connectivity Map in drug discovery and development. Drug Discov Today. 2012. In press
- Fojo, T. and D.R. Parkinson, Biologically targeted cancer therapy and marginal benefits: are we making too much of too little of are we achieving too little by giving too much. Clin Cancer Res. 2010.16(24):5972-80
- J. Arrowsmith. Trial watch: Phase II failures: 2008-2010. Nat Rev Drug Disc. 2011. 10: 328-329
- Eschenhagen, T., et al., Cardiovascular side effects of cancer therapies” a position statement from the Heart Failure Association of the European Society of Cardiology. 2011. 13(1): 1-10
- Cunningham, D., et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004. 351(4): 337-45
- Balko, J.M., et al. Gene expression signatures that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and lung tumors. BMC Genomics. 2006. 7:289
- Ferriss, J.S., et al., Multi-gene expression predictors of single-drug responses to adjuvant chemotherapy in ovarian carcinoma: predicting platinum resistance. PLoS One. 2012. 7(2):e30550
- Kadra, G., Gene expression profiling of breast tumor cell lines to predict therapeutic response to microtubule-stabilising agents. Breast Cancer Res Treat. 2012. 132(3):1035-47
- Balko, J.M., et al. Combined MEK and EGFR inhibition demonstrates synergistic activity in EGFR-dependent NSCLC. Cancer Biol Ther. 2009. 8(6): 522-530
- Haura, E.B., et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010. 16(8): 2450-7
- Feng, X., et al. Pharmacogenomic biomarkers for toxicities associated with chemotherapy. US Pharm. 2012. 37(1)(oncology supplement): 2-7
Biography
Esther P. Black, PhD, is a faculty member at the University of Kentucky, College of Pharmacy and Markey Cancer Center. Dr. Black received a PhD in biomedical science at the University of Florida and completed a postdoctoral fellowship at Duke University with Joseph Nevins, PhD. She is co-Founder of TrackFive Diagostics, Inc.