

Anatomy of a ‘correlate of protection’ for T-cell vaccines
Posted: 28 June 2022 | Thomas Rademacher (Emergex Vaccines) | No comments yet
The use of proprietary vaccine constructs to prime naive CD8+ T-cells via skin dendritic cells and thus enable induction of targeted cellular immunity, establishing tissue-resident CD8+ T-memory cells, is a new vaccination strategy being explored at Emergex Vaccines. Here, Professor Thomas Rademacher, Emeritus Professor of Molecular Medicine – UCL and CEO of Emergex Vaccines, discusses the importance of cellular immunity to protect against viral disease and explains how delivery of virus-specific major histocompatibility complex (MHC) class I epitopes to the skin-resident immune system can translate to the presence of antigen-specific memory T-cells at distal sites. Such tissue-resident T-cells are able to provide frontline defence against invading pathogens and may be an important ‘correlate of immune protection’ (ie, the immune response a vaccine needs to trigger so that an individual is protected from future infectious disease – this correlate differs from immunologic memory).
Cellular immunity and vaccination
The development of vaccines with a strong Type 1 T helper (TH1)-type cellular immune component, such as CD8+ cytotoxic T lymphocytes (CTL), has been limited to the historical observation that only live‑attenuated vaccines (such as yellow fever, smallpox vaccines, etc) appear to generate a CTL response which can afford a lifetime ‘correlate of protection’. Inactivated, virus-like particle and subunit vaccines may only give rise to limited CD8+ T-cell repertoires that differ qualitatively and quantitatively from those generated in natural infection. Class I epitopes from non-living vaccines can only be derived by cross-presentation of peptides generated during class II processing of extrinsic viral proteins in antigen presenting cells. These extrinsic proteins do not represent the entire viral proteome and are overrepresented by the structural proteins making up the viral surface or are limited to protein(s) in the vaccine. DNA and mRNA vaccines could potentially give rise to a limited set of class I peptides from a single protein beyond those generated by cross‑presentation, but no robust evidence for this outcome has yet been published.
Measured CD8+ T-cell responses require enhancement techniques for detection, and it is unclear whether these CD8+ T-cells are cross‑reactive or virus-specific, or above a threshold level required to provide meaningful cellular immune protection – the viral proteins generated by these vaccines and expressed in healthy cells are non-self (foreign) but non-immunogenic, as previously demonstrated in numerous transgenic animal experiments. Generation of class I peptides via cross-presentation is inefficient, stochastic and, in some cases, such as for the Severe Acute Respiratory Syndrome coronavirus 2 (SARS‑CoV-2) spike protein, no class I peptides are actually produced.
It is thought that only one virion is required/permitted to infect a target cell;1,2 therefore, it is hard to see in practice how antibody-based vaccines can be sterilising”
Vaccines that are mainly antibody-inducing have strong CD4+ T-cell components, but, in many cases, were developed as simple anti-toxin immune therapies (eg, tetanus [toxin], diphtheria [toxin] and COVID-19/anti-spike vaccines). Consequently, they do not generate protection against infection nor constrain viral transmission. It is thought that only one virion is required/permitted to infect a target cell;1,2 therefore, it is hard to see in practice how antibody-based vaccines can be sterilising. Their protective efficacy is normally established in lethal challenge experiments using animal models, in which a reduction in viral load is intrinsic to success. These studies, however, have little relevance to real-life transmission characteristics, which are normally governed by exceedingly small inoculum size. The viral ‘bottleneck’ is the number of virions required to transmit from a donor to recipient. Circulating viruses tend to have smaller bottlenecks as they adapt to a population with a bottleneck of one, the final end game. For example, the COVID-19 Delta variant was shown to have a bottleneck approaching one (influenza is estimated at between one and approximately 100), suggesting that the Omicron variant did not evolve as a ‘superior’ transmission cloud. The quantity of Dengue virus in a single mosquito bite is below the level of detection by polymerase chain reaction (PCR).
Most circulating endemic and pandemic viruses are RNA viruses and are thus not actually defined single-genetic entities (unlike DNA viruses) but are an ensemble of quasi-species, the sum of which is called the consensus sequence. COVID-19 virus variants (as defined by a consensus sequence) do not really exist as discrete entities, but merely reflect changes in an experimentally‑measured consensus sequence that remains fixed until conditions for viral population survival changes. The clouds are transmissible, due to the deterministic behaviour of the ensembles (ie, no matter which set of quasi-species is transmitted [founders], the population that evolves in the recipient will match the original donor even for a bottleneck of one). All members of the ensemble work together to create the observed behaviour. Changes in consensus sequence occur at the population level, ie, during the short course of a single infection cycle, a new mutant quasi‑species cannot reach a frequency that would impact the behaviour of the ‘cloud’ as a whole. Cellular immunity has the ability to target common/conserved elements of the quasi-species cloud,3,4 in contrast to vaccine-induced antibodies that predominantly bind to structural proteins exposed on the virion surface (which may differ substantially between quasi-species). In several cases, it was demonstrated that no protein actually existed that matched the consensus sequence for that protein.
Pre-activated naïve CD8+ T-cells can eliminate infected cells before productive infection
“T-cell-specific vaccines can generate an army of CD8+ T-cells that are specific for eclipse-phase/pioneer round antigens5,6”
The eclipse period of a viral infection is an important concept used in immunology and vaccinology to describe the time after a cell has been infected and is able to produce new virions that are transmitted cell-to-cell or released into a fluid compartment. Immediately upon infection, and as soon as the synthesis of viral protein is initiated (completed proteins are not required), the infected cell will generate class I-binding, virus-derived peptide epitopes that mark the cell as foreign. If there are pre-existing CD8+ T-cells already available in the body against these early targets, they can kill the infected cell before it has productive virion release – a fortunate result known as an ‘abortive infection’ and the basis of the anamnestic response in natural immunity. A virus that largely spreads cell-to-cell can hide from antibodies entirely.
Immunological memory in natural infection
Natural immunity relies on continuous low‑grade re-exposure over a lifetime, to periodically re‑prioritise a person’s memory T-cells. The lymphoid system has limited shelf-space and memory cells simply cannot keep being added. After each viral infection, lymph nodes first undergo an expansion of 10–20x in volume and then contract. The massive cull of immune cells during the contraction phase is required to keep the total number of lymph node cells fixed. Many of the T-cells that die are ‘bystander cells’ – cells that were generated to previous infections. Post thymic involution, we also will lose our unique set of naïve T-cells as we age, for they cannot be replaced. Essentially, with each subsequent infection, old memory T-cells, etc, are eliminated. Memory T-cells from previous infections must periodically be ‘recharged’ and moved up the hierarchy of available protection. The latest viral infections, however, have an advantage in that many of the viral antigens get ‘archived’ in afferent lymph node endothelial cells, providing a source of antigens to maintain memory for the last infection.
Protection against a tissue viral infection via CD8+ T-cells is mediated specifically by tissue-resident memory cells. After a natural infection, T-cells will take up residence in tissue niches that are sites for a secondary reinfection (ie, the lungs for influenza, skin for dengue, liver for malaria, etc). To date, the generation of tissue-resident memory T-cells has only been shown to occur after live virus infections and no current, non-living vaccine has been shown to achieve this important correlate of protection.
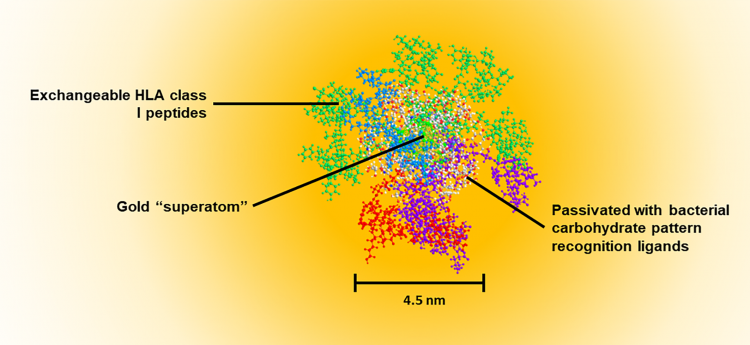
Figure 1: Water-soluble, quantum-confined clusters are self-assembled in a single step to create a three-layered structure. The central gold superatom (102 atoms, 1.6nm diameter) exhibits no plasmon resonance, but does create a 1/d4 energy transfer condition to the metal surface. This process results in quenching of fluorescent molecular dyes and disruption of water/ionic structure/Brownian motion around the particle that leads to deactivation of enzymatic activity at the surface layer of the superatom. This field effect has been shown to result in stabilisation of RNA and peptides on the surface of the quantum-confined cluster gold cores. The orange shading shows the magnitude of the field effect (fluorescent quench) as a function of distance from the metal surface. The second layer (thatch layer) is a passivation layer and renders the particle water-soluble. It consists of bacterial mimetic carbohydrates that are known pattern recognition elements (PRE). This surface ‘thatch’ is recognised by the innate immune system such that the particle resembles a bacterial degradation fragment which is recognised as ‘infectious’. The sugar component of almost all bacterial surfaces consists of alternating residues of β4-N-acetylglucosamine (β4GlcNAc) and β4-N-acetylmuramic acid (β4MurNAc). The latter is just a phosphoenolpyruvate adduct of GlcNAc. Innate recall (anamnestic response) to these surfaces occurs in the absence of inflammation. This condition is critical since T-cell memory does not evolve in the presence of inflammation. In a natural infection, activation of naïve T-cells by dendritic cells will occur before systemic inflammation occurs – ie, priming occurs before physical symptoms. The PRE can stimulate NKT cells to promote ‘signal 0’ which can substitute for CD4 T cell help. The outer layer contains class I-binding peptides that are attached to the passivated core particle via a thiol–gold linkage. In extracellular fluid, reducing compounds such as glutathione are not at a sufficient concentration to induce Murray‑Place exchange and release the peptide from the particle. However, cells normally contain 5 mM glutathione, and this results in almost immediate release of peptides from the particle upon entry into the cytoplasm after transiting the plasma membrane. The passivated layer results in almost exclusive uptake of the particles by antigen presenting cells, as determined by monitoring gold uptake of cells using cytometry by time of flight (CytoTOF) mass spectrometry. All peptides are attached in the C-to-N orientation with a linker on the N terminal. This linker provides the recognition motif for translocation of the peptide to the endoplasmic reticulum where the peptide is then inserted C terminal end into a vacant class I molecule. The N terminal trimming proteases then cleave the linker resulting in a peptide of defined length (9–15 mer), identical to those empirically found in class I MHC expression ligandomes. The final vaccine construct is ~4.5nm in diameter which permits passage through collagen network in the skin (10nm), uptake to lymphatics (20nm), permeation through vascular pores (6–8nm) or free clearance from kidneys (5–6nm). This size contrasts with lipid encapsulated vaccine vehicles (>50nm) that can only access the sinusoids of the liver (150nm).
Vaccination to generate tissue-resident memory CD8+ T-cells
T-prime vaccines can generate blood-circulating CD8+ T-cells that have the clonal imprinting to enable them to become tissue-resident memory T-cells. However, they will only make this transformation after a natural infection; that is, infection at tissue level will take hold before the circulating T-cells can home to affected tissues and have any impact on initial viral load.
What is critical to managing the disease is to have the tissue-resident T-cells present ‘prior’ to the infection. T-priming vaccines can give rise to a T-cell army (primed) ready to act on cells as soon as they are infected. In contrast, ‘prime-trap’ vaccines both prime the T-cell army and then provide the stimulus for homing of the T-cells to the tissues, thus converting them to tissue-resident cells (the homing ‘trap’) in the absence of infection. These types of vaccines have demonstrated feasibility in animal models of influenza and malaria. Emergex currently has such prime-trap CD8+ T-cell vaccines in clinical trials for dengue and COVID-19. The candidate vaccines are composed of synthetic peptides attached to a gold nanoparticle carrier system (Figure 1) with individual peptide clusters combined to produce the vaccine (Figure 2).
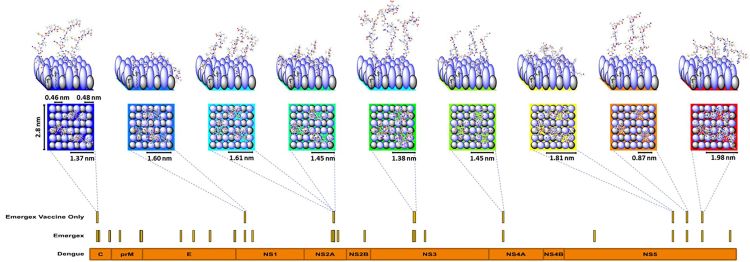
Figure 2: The vaccine particle construct shown in Figure 1 can be deconstructed and represented as a square chip with sides of 2.8nm with 42 ligands consisting of the passivated pattern recognition elements and the class I peptides for delivery to cells. On average each particle contains three copies of each class I peptide, as shown in the Figure. It also shows the process for generating a vaccine candidate from a Dengue-infected cell of specified human leukocyte antigen (HLA) type (each human cell will contain up to six main HLA alleles, ie, 2 HLA-A, 2 HLA-B, 2 HLA-C and some other alleles such as HLA-E, etc). Specific alleles can be grouped as supertypes, expanding the recognition of any peptide to multiple alleles contained within a supertype family. Class I peptides are recovered from infected cells as a time course after infection to determine eclipse-phase epitopes and those generated up to the time of virion release and prior to any cytopathic events. This feature is called the peptide-MHC class I (pMHC) expression library for a given set of supertypes. As these cells do not express class II, the only recovered peptides must be bound by class I molecules and generated by the cell from growing peptides prior to formation of fully post-translationally modified proteins. This contrasts with cross-presentation in class II-expressing antigen presenting cells, in which fully processed intact proteins are proteolytically degraded. Each of the recovered class I peptides can be assigned to either the self-repertoire or matched to sequences unique to the infecting virus (labelled ‘Emergex’ in the figure). These sequences can be mapped to the dengue proteome as shown in the orange bar. From this ‘ligandome’, a selection of peptides is made for the final vaccine shown as ‘Emergex vaccine only’. Each of these peptides can then be attached to a different chip and the chips combined to form a vaccine construct that will specifically activate naïve T-cell clones which recognise the peptides. There are potential ‘holes’ in the naïve T-cell repertoire to dengue for any individual; but across a population, all the peptides should be immunogenic. The use of multiple peptides generates a T-cell army. T-cells are limited serial killers (approximately 100 kills), but to kill an infected cell, multiple interactions are required (at least five), so a swarm is required. In practice, all the ‘chips’ can be made simultaneously in the same synthesis process.
The first aim of T-cell vaccination is to generate an army of CD8+ T-cell clones against specific viral antigens that occur during the eclipse phase of a viral infection. This set of specificities is defined experimentally at target-cell level and not the immune level; that is, T-cell specificities are only relevant if an infected cell actually expresses the epitope (off-target T-cells generated are of no use). The repertoire of peptides present in class I molecules from infected cells is called the ‘target‑cell MHC expression library’ or ’ligandome’. To achieve the virus-homing trap, non-self, but non‑immunogenic, seeding of viral class I peptides occurs during a second vaccination. The initial vaccination primes skin dendritic cells (DCs) to activate naïve T-cells in the draining lymph nodes, which recognise class I complexes binding the vaccine peptide. This results in the expansion of T-effector cells (their frequency increasing by several orders of magnitude) and circulating T-memory cells that have tissue‑homing potential. The second vaccination targets distal tissue DCs that can present antigens to the circulating T-memory cells, which then evolve into tissue‑resident T-memory cells. In addition to direct uptake of the vaccine by DCs, both the prime and trap functions can be mediated by a process called ‘cross-dressing’ (or trogocytosis), ie, the acquisition of pre-formed peptide-MHC complexes from donor cells to recipient cells. This process generates pseudo‑infections (in contrast to direct infection) of DCs at, and distal from, the vaccination site. For example, in the skin, the donor cells would be keratinocytes and the recipients the resident dendritic/Langerhans cells. In blood, monocytes appear to be the main donor.
Conclusion
T-cell-specific vaccines can generate an army of CD8+ T-cells that are specific for eclipse-phase/pioneer round antigens.5,6 Some of these T-cells will have gained, by asymmetric division, the capacity to home to tissues and set up tissue-resident CD8+ T-cell armies that are critical to preventing infections – especially in the lungs to protect against respiratory infections or in the skin to protect against arbovirus infection. This cellular army can be activated either by subsequent natural infection or by using vaccine-dispensed epitope distribution to tissue sites to generate protective resident T-memory cells, which (in the absence of any infection) may result in a beneficial abortive infection.
A fully referenced version of this article can be found at: www.emergexvaccines.com/media-centre
About the author
Professor Thomas W. Rademacher, MD, PhD is Emeritus Professor of Molecular Medicine at University College London Medical School and CEO of Emergex Vaccines. Professor Rademacher was co-Founder of the Oxford Glycobiology Institute and subsequent spinout Oxford GlycoSciences PLC. He was also Founder, Chairman and CEO of the first nanomedicine company, Midatech Ltd.
References
- Watanabe Y, Arai Y, Kawashita N, Ibrahim MS, Elgendy EM, Daidoji T, et al. Characterization of H5N1 influenza virus quasispecies with adaptive hemagglutinin mutations from single-virus infections of human airway cells. J Virol. 2018;92(11):e02004-17.
- Zwart MP, Hemerik L, Cory JS, de Visser JA, Bianchi FJ, Van Oers MM, et al. An experimental test of the independent action hypothesis in virus-insect pathosystems. Proc Biol Sci. 2009;276(1665):2233-42.
- Viruses are models for embracing diversity. Nat Microbiol. 2018;3(4):389.
- Ternette N, Block PD, Sanchez-Bernabeu A, Borthwick N, Pappalardo E, Abdul-Jawad S, et al. Early Kinetics of the HLA Class I-associated peptidome of MVA.HIVconsv-infected cells. J Virol. 2015;89(11):5760-71.
- Swadling L, Diniz MO, Schmidt NM, Amin OE, Chandran A, Shaw E, et al. Pre-existing polymerase-specific T cells expand
in abortive seronegative SARS-CoV-2. Nature. 2021;601(7891):110-7. - Apcher S, Daskalogianni C, Lejeune F, Manoury B, Imhoos G, Heslop L, et al. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc Natl Acad Sci U S A. 2011;108(28):11572-7.
Issue
Related topics
Biologics, DNA, Drug Development, Drug Targets, mRNA, Research & Development (R&D), t-cells, Vaccine Technology, Vaccines, Viruses
Related diseases & conditions
Coronavirus, Covid-19, Dengue, Severe Acute Respiratory Syndrome (SARS)





