

GPCRs: Cell based label-free assays in GPCR drug discovery
Posted: 20 August 2013 | | No comments yet
G protein-coupled receptors are one of the major classes of therapeutic targets for a broad range of diseases. The most commonly used assays in GPCR drug discovery measure production of second messengers such as cAMP or IP3 that are the result of activation of individual signalling pathways. Such specific assays are unable to provide a holistic view of the cell response after GPCR activation.
This is now changing as label-free technologies and assays on whole cells have been developed that are unbiased towards the specific downstream pathways and capture the integrated cell response. In this mini-review, we focus on the application of one of these technologies, namely resonant waveguide grating (RWG) for measurements of dynamic mass redistribution (DMR) in intact cells upon GPCR activation. Since the technology is sensitive and non-invasive, it is applicable to most cell types, including primary cells with native receptor expression levels. We discuss how DMR assays have become an important component of GPCR drug discovery screening cascades and may have the potential to improve the ability to predict if compounds will be efficacious in vivo.
G protein-coupled receptors (GPCRs) are among the most important target classes within the pharmaceutical industry1. Of the currently marketed small-molecule medicines, approximately 30 per cent target GPCRs2,3. Historically, ligand interactions with GPCRs have been analysed using binding assays providing both affinity and kinetic data. This technology did not however take into account functional aspects of the ligands (for example agonism, inverse agonism, allosteric effects or signalling pathway). Many downstream signalling assays like detection of cAMP, IP3, Ca2+-flux, β-arrestin recruitment or ERK1/2 phosphorylation have therefore been applied extensively, expanding the knowledge of functional aspects of ligands4 (Figure 1). These assays have also made it possible to search for new drugs on orphan receptors. The expanding knowledge around signalling through different G-proteins, G-protein independent signalling and signal bias, have pinpointed the limitations with assays based on detecting specific intracellular messengers along one single pathway5. However more recently, label-free biosensor technologies such as surface plasmon resonance (SPR), resonant waveguide grating (RWG) and cell impedance spectroscopy (CIS) have been developed and applied in drug discovery providing an opportunity for the revelation of pharmacology of greater physiological and disease relevance than before, including simultaneous capturing of signalling via multiple pathways6.
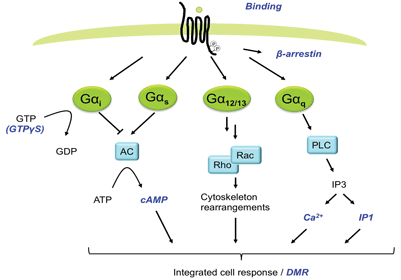
Figure 1
In this review, we focus on the application of RWG for measurements of DMR in intact cells upon GPCR activation. DMR is a consequence of morphological changes and changes in distribution of cellular components after receptor stimulation. This type of readout is still somewhat of a black box, but consists of elements such as protein recruitment, receptor internalisation, reorganisation of the cytoskeleton, as well as altered cell adhesion (Figure 2). Activation of all four major GPCR coupling classes (Gαi/Gαo, Gαs, Gαq/Gα11 and Gα12/Gα13) can be captured with this technology7, something which is unachievable with most other platforms. Initial studies suggested that signalling via different G-proteins result in class-specific DMR kinetic profiles8. Subsequent studies have however revealed that kinetic signatures are cell type specific and the picture is therefore more complex than initially suggested. Nonetheless, DMR provides a G-protein unbiased and pathway sensitive technology as a powerful complement to single pathway technologies in the GPCR assay toolbox.
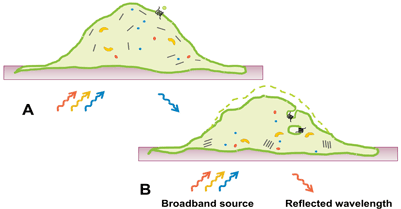
Figure 2
Examples of applications in the drug discovery process
We and others have applied cell based label-free DMR GPCR assays for different purposes in various phases of early drug discovery (Figure 3).
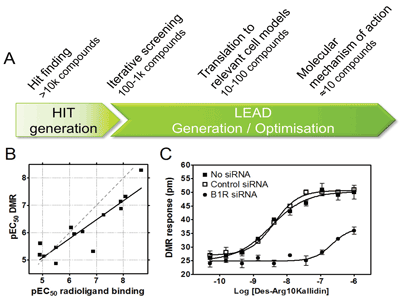
Figure 3
Hit finding
Ligands can be biased with respect to signalling via different pathways, thereby making the pharmacology observed highly dependent on the assay selected9. The potential benefit of capturing signalling via multiple pathways and thereby cover compounds with biased signalling makes DMR an attractive choice for identification of novel chemical starting points. However, only a limited number of large DMR screens have been published so far, likely due to the relatively recent introduction of the technology, high plate costs and throughput challenges including detection of slow responses using kinetic reading. One published example is a 100k screen for muscarinic M3 antagonists10. In this screen, a low hit rate consistent with many other HTS formats was observed. The output did not suffer from a high rate of false positives. Most importantly, unique hits that were not picked up in a corresponding Ca2+ FLIPR® screen were identified and confirmed in a binding assay. Thus, this example illustrates the potential of DMR to successfully deliver a relatively clean but unbiased HTS output including chemistry which is not captured by single pathway technologies.
Iterative screening
Our own most common application of DMR is as an integral part of screening cascades within the lead generation phase, either as the primary structure-activity-relationship (SAR) driving assay or as an orthogonal assay. We define an orthogonal assay as an assay that measures activity on the same target as the SAR driving assay but with an independent technology, which is applied to strengthen confidence in potency / efficacy measures from the SAR assay. In this context, DMR assays are of particular value where other screening friendly assay alternatives are limited, for example, for GPCRs such as GPR55 which signals via the Gα12/13 pathway7, for receptors lacking appropriate radioligands, or for antagonist screens on Gαi coupled receptors. In addition, DMR assays are often technically straightforward to establish.
Some concerns about the biological validity of DMR assays to drive SAR have been raised due to the undefined / black box nature of the readout. However, this can be mitigated by control experiments to confirm target dependence such as demonstrating absence of compound effects in comparable cell lines lacking the target. It is also crucial to establish early correlations between DMR and pathways specific or binding assay platforms to interrogate the chemical series of interest with respect to potential signal bias. Such a correlation, which is a prerequisite for application in screening cascades, has been established both for overexpressed and endogenously expressed receptors and is our own experience as exemplified by the bradykinin B1 receptor (Figure 3B).
Translation to biologically relevant cell models
It is often a key challenge within early drug discovery projects to establish a link between assays using recombinant reagents and more biologically or disease relevant cell models. For example, overexpression of receptors can shift both the potency and efficacy of compounds (often overestimate) due to spare receptors, thus not reflecting the pharmacology in a native tissue11. Moreover, native tissues also ensure the right G-protein composition and expression relative to receptor levels, and this may even be tissue-specific. Switches in G-protein coupling have frequently been seen for receptors expressed in different cell lines, but are not limited to engineered cells12. For example, endogenous dopamine D1/D2 receptor responses couples to different G-proteins in different cell types12 highlighting the importance of selecting disease relevant cells also for in vitro screening.
The high sensitivity of DMR assays enable measurements of signalling after activation of endogenously expressed GPCRs. Most importantly, since DMR assays do not require genetic manipulation of cells, even primary cells from patients can potentially be employed. An example using non-engineered cells is a 1280 compound screen using human epidermoid carcinoma A431 cells endogenously expressing histamine H1 and β2 adrenergic receptors13. With respect to cells from native tissue, it has also been shown that DMR has the potential to detect prostaglandin E1 (PGE1) activation of EP2/EP4 prostanoid receptors in primary human keratinocytes7. In our own lab, we have shown that activation of Bradykinin B1 receptors (B1R) can be monitored in primary human preadipocytes and that the DMR response is target mediated as it can be reduced by knock down of B1R using siRNA, demonstrating specificity of the response (Figure 3C). In summary, DMR assays commonly have sufficient capacity and sensitivity to provide a relatively high throughput link between assays on recombinant reagents and disease models to ensure relevant potency, efficacy and coupling mechanisms of hit or lead series early on.
Molecular mechanisms of action studies
Molecular mechanism of action (MMOA) is defined here as the interaction between a compound and its target, resulting in a specific response3. These specific molecular interactions link structure to function to provide a biologically meaningful response. MMOA studies are important for selection of functionally relevant compound series and are generally performed on small compound sets during late lead generation and lead optimisation.
A key application in this context is characterisation of signalling pathways in physiologically relevant cells. The relative contribution of different pathways can be addressed by co-treatment with G-protein / pathway specific blockers such as pertussis toxin (Gαi), cholera toxin (Gαs) and YM254890 (Gαq)7 with subsequent measurement of the remaining signal after agonist stimulation. When inhibitors are not available, dominant negative G-protein forms or siRNA can be used as alternative tools7. Bock et al have taken mechanistic studies one step further by taking advantage of DMR and dualsteric ligands (i.e. an orthosteric ligand physically linked to an allosteric modulator) for elucidating the dependence of promiscuous G-protein activation upon conformational rearrangements within the extracellular domain14.
With respect to other MMOA applications, it is technically possible to assess conformational mechanisms (e.g. surmountable / insurmountable, competitive / non-competitive compounds) but here DMR assays do not offer any obvious benefits compared to classical techniques. In contrast, for determination of efficacy, DMR assays have the potential to provide relevant measures over the range from full agonists via partial agonists to inverse agonists12. This is of particular value in cases where it is possible to measure on native(like) cells with endogenous receptor expression levels.
Concluding remarks
Plate-based label-free technologies like RWG that can be employed for cell based applications such as DMR assays have been available for drug discovery for approximately five years and the number of applications is still growing. Since the technology is sensitive, non-invasive and able to detect signalling via multiple pathways, it can be applied on disease relevant cell systems. These properties offer the possibility to use the same type of readout for hit finding, iterative screening, (including species and target selectivity), and disease relevant cell models and may help to improve the ability to predict if compounds will be efficacious in vivo.
In the future, we expect label-free cell based assays with sufficient throughput for early drug discovery to be commonly applied in compound screening on disease relevant cells, generating valuable chemical starting points to be developed into tomorrow’s medicines.
References
- Ma P, Zemmel R. Value of novelty? Nat Rev Drug Discov. 2002 Aug;1(8):571-2.
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002 Sep;1(9):727-30
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011 Jun 24;10(7):507-19
- Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012 Mar;33(3):372-84
- Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011 Mar;17(3):126-39
- Cooper MA, Halai R. What is label-free screening and why use it in drug discovery? European Pharmaceutical Review. 2012;17(6):51-3
- Schroder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, et al. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol. 2010 Sep;28(9):943-9
- Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007 May-Jun;55(3):314-22
- Kenakin TP. Cellular assays as portals to seven-transmembrane receptor-based drug discovery. Nat Rev Drug Discov. 2009 Aug;8(8):617-26
- Dodgson K, Gedge L, Murray DC, Coldwell M. A 100K well screen for a muscarinic receptor using the epic label-free system–a reflection on the benefits of the label-free approach to screening seven-transmembrane receptors. J Recept Signal Transduct Res. 2009;29(3-4):163-72
- Kenakin T, Christopoulos A. Analytical pharmacology: The impact of numbers on pharmacology. Trends Pharmacol Sci. 2011 Apr;32(4):189-96
- Peters MF, Vaillancourt F, Heroux M, Valiquette M, Scott CW. Comparing label-free biosensors for pharmacological screening with cell-based functional assays. Assay Drug Dev Technol. 2010 Apr;8(2):219-27
- Tran E, Ye F. Duplexed label-free G protein–coupled receptor assays for high-throughput screening. J Biomol Screen. 2008 Dec;13(10):975-85
- Bock A, Merten N, Schrage R, Dallanoce C, Batz J, Klockner J, et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat Commun. 2012;3:1044
Biographies
Dr. Niklas Larsson obtained his PhD 1999 in Cell and Molecular Biology at Umeå University, Sweden. Since 2000, he has been working with early drug discovery projects at AstraZeneca (Mölndal, Sweden) in different positions such as Project Leader, Team Leader and currently as Associate Principle Scientist within the Discovery Sciences function. His scientific focus is drug discovery and molecular pharmacology of GPCRs.
Linda Sundström has over 10 years’ experience in GPCR targeted drug discovery. She received an MSc in Chemical Engineering from Lund Institute of Technology, Sweden during which she completed a thesis work at the Institute of Biotechnology, University of Cambridge, UK. She is currently an Associate Principal Scientist at AstraZeneca Discovery Sciences, Sweden where her primary focus is on GPCR pharmacology and applications of cellular assays on GPCR targets in early drug discovery.
Dr. Erik Ryberg obtained his PhD in Biomedical Science at the University of Aberdeen, UK in 2009. He has been working with GPCR related drug discovery at AstraZeneca for over 10 years in different positions. He is currently an Associate Principal Scientist within the Cardiovascular and Metabolic Diseases function with primary focus on molecular pharmacology of GPCRs in different phases of drug discovery.
Lovisa Frostne joined AstraZeneca (Mölndal, Sweden) in 2003 after obtaining her MSc in Molecular Biology at the University of Gothenburg, Sweden. She completed her thesis work at the Institute for Molecular Bioscience at the University of Queensland, Australia. During her 10 years in AstraZeneca, her work has been focused around GPCR pharmacology in early drug discovery. She now holds a position as Team Leader in the Bioscience department within the Respiratory, Inflammation and Autoimmune iMed.
