

Connecting HCS to CNS drug targets
Posted: 2 February 2006 | | No comments yet
HCS has been implemented as a key technology to address complex biology associated with CNS drug targets. This review will describe a new generation of HCS assays including multiplexed HCS assays with biochemical markers, novel techniques for studying receptor internalisation and the application of HCS to neural network cultures that have facilitated CNS drug discovery.
High Content Screening (HCS) enables the measurement of diverse subcellular events by integrating whole cell fluorescent assays with automated image acquisition and processing algorithms. Whilst a number of HCS screening platforms are currently available including the Arrayscan (Cellomics), IN Cell Analyzers 1000 and 3000 (GE Healthcare) and the Opera (Evotec Technologies), only a handful of companies are currently utilising this technology to aid drug discovery. This may be partly attributed to the significant costs associated with HCS instrumentation coupled with the demands on IT resources to provide the relevant storage and data mining capabilities. On the other hand, a number of companies have invested heavily to incorporate HCS as an integral part of the drug discovery process and have made significant progress in this developing area.
The first HCS assay was reported in 1998 by scientists from Cellomics and Merck Research Laboratories, describing a nuclear translocation assay to detect NFκB translocation from the cytoplasm to the nucleus in HeLa cells (Ding et al., 1998). The assay was performed in 96-well format and detected endogenous levels of NFkB within nuclear and cytoplasmic cellular compartments, following stimulation with the pro-inflammatory cytokines, TNF-α and IL-1. Using this assay, it was possible to determine IC50 values for compounds blocking nuclear translocation of NFκB. As well as detecting transcription factor activation, HCS assays and algorithms have since been expanded to measure G-protein coupled receptor (GPCR) signalling including receptor internalisation, apoptosis, cytotoxicity, cell viability and neurite outgrowth. The translocation HCS assay was developed further to facilitate quantification of proteins translocating from the plasma membrane to the cytosol and vice versa. Such assays represent standard HCS techniques that are now relatively easy to implement, provided a suitable antibody to the protein of interest can be identified, a relevant cell line exists or can be developed and an algorithm to quantify the change is available.
Simple modification of standard HCS techniques can result in more specialised assays. This review will describe the development of novel HCS assays that have been implemented within the Terlings Park site at Merck to facilitate CNS drug discovery. These so-called second generation HCS assays include high information assays with triple parameter readouts, HCS assays to measure native receptor activation and application of HCS to primary neuronal network cultures.
Nuclear Translocation Assays using Epitope Tagging
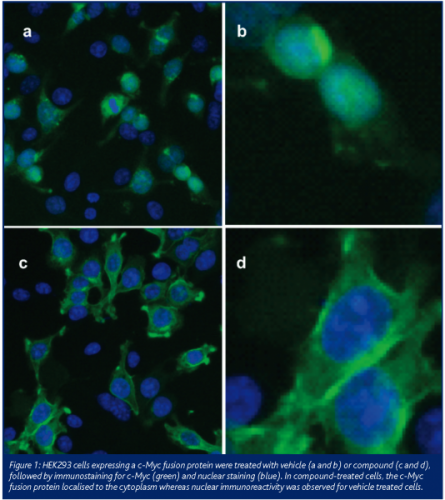
Conventional nuclear translocation assays such as the one described by Ding and co-workers (1998) require a specific antibody to the protein of interest to determine the relative fluorescence intensity in the nucleus versus the fluorescence intensity in a sample area of the cytoplasm at a given time. This method is limited by the availability of suitable antibodies that do not cross-react with non-specific proteins. More recently, nuclear translocation assays using GFP fusion proteins have been described for transcription factors and protein kinases, now known as Redistribution Screening Technology (Almholt et al., 2004). This approach enables the visualisation of protein translocation in live cells, providing a suitable nuclear label is used that is spectrally distinct from GFP. Coupled with recent advances in confocal imaging capabilities, the ability to track protein translocation in live cells represents a powerful technique for studying intracellular protein movement. A key advantage of this approach is that it is not antibody dependent, however, GFP itself can affect the dynamics of protein translocation in certain cases. Recently, Invitrogen introduced LumioTM, an in-cell labelling kit enabling detection of recombinant proteins in live cells, using fluorescence microscopy (Griffin et al., 1998). LumioTM consists of a tetracysteine tag which is fused to the gene of interest and the expressed fusion protein can be visualised upon addition of a labelling reagent – LumioTM green or LumioTM red. The particular motif used is rarely seen in naturally occurring proteins and benefits from being only six amino acid residues long and is therefore unlikely to interfere with the biological activity of the protein in question. HaloTagTM (Promega) works in a similar manner to LumioTM and can be used to visualise proteins in both live and fixed cells. An alternative approach that may be more cost-effective is to engineer cell lines where the protein of interest is fused to one or more small non-fluorescent proteins with low levels of endogenous expression. We have successfully used this approach to study nuclear translocation of a protein for which no suitable antibody was commercially available but by fusing the protein of interest to several c-Myc molecules, the protein was visualised using antibodies targeting this epitope tag. By tagging proteins with a generic protein such as c-Myc, the identification of a specific antibody is bypassed and chances of developing a successful HCS assay are increased if a well-characterised tag is used. Like GFP, the use of epitope tagging to track protein movement within cells does require the necessary experiments to exclude the possibility that the protein tag itself interferes with translocation and that the tag is not expressed endogenously or is expressed at low levels within cells. Whilst this approach is limited to end-point assays, advantages over GFP include uniform signal generation across cells as levels of GFP expression can vary on a cell by cell basis. This is a potential problem if the cells have been through a number of passages prior to experimentation.
Multiplexed HCS assays
Multiplexed HCS assays include those where a combination of fluorophores emitting at different wavelengths are used to track the movement of more than one protein in the same well. Additionally, HCS assays are amenable to being multiplexed with other biochemical assays such that data is obtained on multiple parameters using the same cell sample. This allows direct comparisons to be made using, for example, IC50 values obtained from multiple targets within the same cell population and minimises inter-experimental variation. We have used this approach to dissect compound mechanism of action in addition to ascertaining the intracellular potency of compounds in a single assay. This was made possible by harvesting the cell-conditioned media prior to fixation and quantifying the effects of compounds on the levels of secreted peptide within this fraction. Meanwhile, the cell monolayer was fixed and immunocytochemistry performed on the same cell population to measure compound effects on nuclear translocation of another protein.
Such multiplexed assays allow streamlining of screening cascades as a single multiplexed HCS assay may be able to replace other biochemical assays with the added advantage of generating imaging data. The increasing use of HCS assays during all phases of drug discovery, but especially during lead identification and optimisation processes, could see a decline in the use of traditional biomolecular, cell-free screening with scientists using an HCS assay as their primary screen. Following high throughput screening (HTS) campaigns, cell-free assays are traditionally positioned as the frontline screen for evaluating hits and have been used to confirm the specificity of a compound and target interaction. In certain cases, these assays could become increasingly redundant as whole cell imaging assays evolve and are multiplexed with other biochemical markers. For certain target classes, recent trends suggest increased emphasis on determining intracellular compound activity prior to confirming target specificity. This is made more possible by the recent introduction of automated cell culture systems such as SelecT (The Automation partnership) that can now address the bottlenecks associated with cell supply.
Novel approaches to receptor internalisation
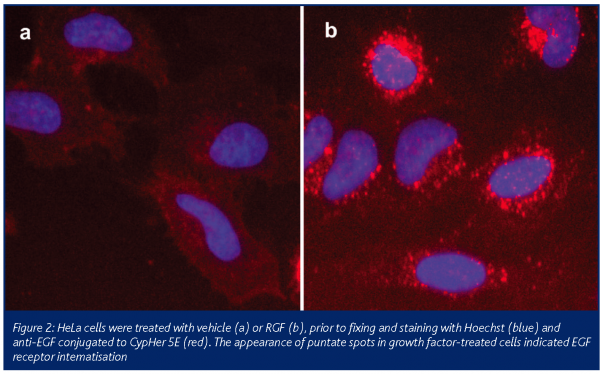
A number of screening formats exist for measuring GPCR activation. Traditional HCS assays to detect GPCR activation typically measure receptor internalisation, following agonist stimulation in a cell line expressing the GPCR of interest. Recently, technologies such as Transfluor Technology (Xsira Pharmaceuticals) have been used successfully in HTS campaigns to identify compounds modulating receptor activation by measuring β-arrestin recruitment. This approach requires cell lines expressing the GPCR of interest in addition to GFP-labelled β-arrestin. However, the use of modified cell lines overexpressing GPCRs can result in higher levels of constitutive receptor internalisation under basal conditions. This in turn results in a smaller response upon ligand activation. Also, direct antibody-based approaches to measuring receptor internalisation are limited by the availability of suitable antibodies and the signal obtained is often small, requiring enhancement for detection purposes. We have investigated a technique using a pH-sensitive cyanine dye that only fluoresces in acidic conditions, called CypHer 5E (GE Healthcare, Adie et al., 2003). Typically, upon ligand binding, GPCRs and receptor tyrosine kinases (RTKs) internalise into acidic endosomes and are either recycled back to the plasma membrane or degraded. Using the EGF receptor as a model system, we labelled antibodies against EGF with CypHer 5E and measured endogenous receptor internalisation in HeLa cells following growth factor stimulation. Using this technique we could show dose and time dependent EGF receptor internalisation. This is the first demonstration of the use of CypHer 5E to measure the internalisation of endogenously expressed receptors. Moreover, the CypHer 5E signal is amplified due to the increased number of labels conjugated to the EGF antibody and therefore the signal is further enhanced compared with traditional antibody-based approaches. As well as applying this technique to unmodified cell lines, this approach may be used to study receptor activation in primary cultures.
Applying HCS to neuronal network cultures
Conventional HCS assays use unmodified cell lines such as HeLa or cell lines engineered to express the desired proteins in host cells such as HEK293 and U2OS. For CNS targets, neuroblastoma cell lines such as SH-SY5Y have been used successfully in high throughput functional assays measuring neuritogenesis, proliferation and differentiation (Simpson et al., 2001). Primary cell cultures are also amenable to both end-point and kinetic HCS (Chan et al., in press, Richards et al., 2006), providing cells adhere for the duration of the assay and the monolayer contains spatially separated cells to facilitate image analysis. We have developed HCS assays using primary cortical cultures established from rat brains. These cultures contain a mixture of cell types interspersed between a meshwork of crosslinking neurites, thus presenting considerable challenges for image analysis using existing algorithms. As HCS is applied to more complex cell cultures, the need for alternative algorithms has become evermore apparent. This is now being addressed by the emergence of new third party image analysis software offering tailor-made solutions to analyse more complex cell responses. The ability to image neuronal network cultures in a 96-well format and incorporate this into a drug screening cascade enables the generation of biologically significant data using physiologically relevant cells. Apart from limitations with existing algorithms, HCS using primary cultures can be acutely sensitive to culture conditions and this can affect data generation. From our experience, when using neuronal preparations, the effects of seeding density, days in culture, quality of plates, serum supplements and media changes can influence biological responses and affect image analysis. Standardisation of culture techniques is pivotal when imaging more complex cell culture systems.
In summary, HCS is viewed by some scientists to provide a potential solution to bottleneck areas in drug discovery due to the ability to perform simple experiments yielding high information, multi-parameter data from a single cell sample. In addition, HCS has the potential to be applied at all stages of drug discovery ranging from target validation through to lead optimisation and safety assessment. Whilst HCS has begun to make an impact on drug discovery, significant developments in areas such as algorithm development and software bio-applications are now required as scientists begin to utilise this technology to its full extent. Such high information assays, as well as capturing key biology, generate multiple datasets, which presents another key challenge for data analysis and interpretation in addition to data storage and mining issues. The contribution of HCS to enzymes, especially kinase screening programmes and as part of deconvoluting hits following blackbox or phenotypic screening approaches was beyond the scope of this article but represent other equally important areas for the application of HCS. Furthermore, new advances in reporter tags and the possibility of studying FRET interactions using HCS may evolve into the next generation of HCS assays.
Acknowledgments
I wish to thank members of the Automated Imaging and Electrophysiology group who contributed to the work described in this review by performing experiments and participating in discussions. I especially wish to thank Robert Newman for providing Figure 2.
References
- Ding, G.J.F., Fischer, P.A., Boltz, R.C., Schmidt, J.A., Colaianne, J.J., Gough, A., Rubin, R.A. and Miller, D.K. (1998). Characterization and quantitation of NF-?B nuclear translocation induced by interleukin-1 and Tumour Necrosis Factor-a. Journal of Biological Chemistry 273 (44): 28897-28905.
- Almholt, D.L.C., Loechel, F., Nielsen, S.J., Krog-Jensen, C., Terry, R., Bjorn, S.P., Pedersen, H.C., Praestegaard, M., Moller, S., Heide, M., Pagliaro, L., Maso, A,J., Butcher, S. and Dahl, S.W. (2004). Nuclear export inhibitors and kinase inhibitors identified using a MAPK-activated protein kinase 2 redistribution screen. Assay and Drug Development Technologies 2 (1): 7-20.
- Griffin, B.A., Adams, S.R. and Tsien, R.Y. (1998). Specific covalent labelling of recombinant protein molecules inside live cells. Science 281: 269-272.
- Adie, E.J., Francis, M.J., Davies, J., Smith, L., Marenghi, A., Hather, C., Hadingham, K., Michael., N.P., Milligan, G. and Game, S. (2003). CypHer5: a generic approach for measuring the activation and trafficking of G protein-coupled receptors in live cells. Assay and Drug Development Technologies 1(2): 251-259.
- Simpson, P.B., Bacha, J.I., Palfreyman, E.L., Woollacott, A.J., McKernan, R.M. and Kerby, J. (2001). Retinoic acid evoked-differentiation of neuroblastoma cells predominates over growth factor stimulation: an automated image capture and quantitation approach to neuritogenesis. Anal. Biochem. 298(2): 163-169.
- Chan, G.K.Y., Richards, G.R., Peters, M. and Simpson, P.B. (2006). High content kinetic assays of neuronal signalling implemented on BD Pathway HT. Assay Drug Dev. Tech. 4: in press
- Richards, G.R., Jack, A.D., Platts, A. and Simpson, P.B. (2006). Measurement and analysis of calcium signalling in heterogeneous cell cultures. Methods Enzymol. – Automated Microscopy Screening (J. Inglese Ed), Elsevier Press, in press.
