

Bio-mimetic chromatography to predict drug distribution in vivo
Posted: 29 October 2010 |
A major concern for the pharmaceutical industry is the high attrition rate (>90 per cent) of potential drug molecules failing during late stages of the drug discovery process. This may be due to lack of efficacy in the clinic, unexpected side effects or unfavourable pharmacokinetics. There is a need for new tools to predict the in vivo distribution of discovery compounds earlier in the drug discovery process, thus reducing the investment in molecules failing unexpectedly in later, more expensive stages of the drug development. There are many in silico prediction tools for estimating pharmacokinetic and toxicological effects of compounds based on their chemical structures or physico-chemical properties…
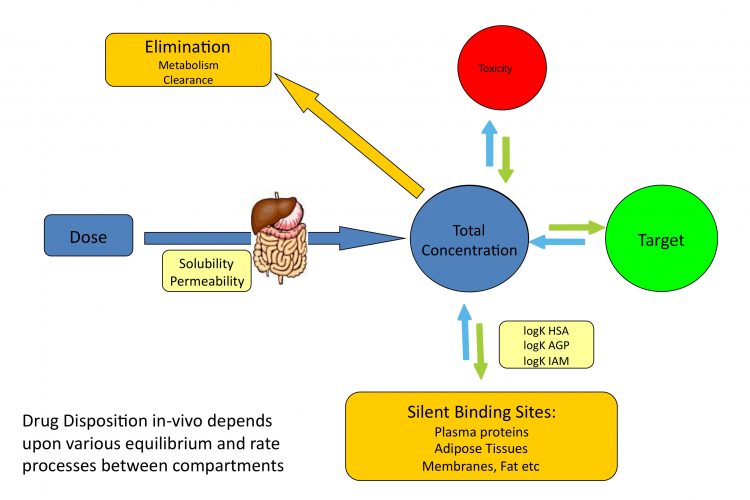
Figure 1 Distribution of drugs in vivo depends on various equilibrium and rate processes between compartments
A major concern for the pharmaceutical industry is the high attrition rate (>90 per cent) of potential drug molecules failing during late stages of the drug discovery process. This may be due to lack of efficacy in the clinic, unexpected side effects or unfavourable pharmacokinetics. There is a need for new tools to predict the in vivo distribution of discovery compounds earlier in the drug discovery process, thus reducing the investment in molecules failing unexpectedly in later, more expensive stages of the drug development. There are many in silico prediction tools for estimating pharmacokinetic and toxicological effects of compounds based on their chemical structures or physico-chemical properties1,2.
However, these models perform poorly when novel molecules with properties dissimilar to those used to train the model are evaluated3. Costly in vivo animal experiments have also proved to be less than satisfactory in accurately predicting the behaviour of the compounds in humans4,5.
Bio-mimetic chromatography provides a simple, inexpensive means to evaluate interactions between compounds and relevant human proteins and phospholipids6 and may be applied very early in the drug discovery process. Utilising this data in predictive models of in vivo drug distribution can aid the identification of potential candidate molecules that will be efficacious at low dose, and consequently less likely to induce toxic side effects, thus reducing the likelihood of late stage attrition. Additionally, an understanding of the molecular attributes contributing to in vivo distribution may help to understand the clinical variation of efficacy and pharmacokinetics, enabling the design of more efficient drug molecules.
Free concentration of drug molecules in plasma and tissues drives in vivo potency
From the early work of Brodie7 on the free drug hypothesis, we know that only the free/ unbound drug in the aqueous compartments of plasma and tissues is available to interact with the target enzyme/protein or receptor. A high proportion of the administered drug binds to silent binding sites and may interact with other off target enzymes and receptors potentially causing side effects and/or toxicity issues. Figure 1 demonstrates how the free drug is in equilibrium with the target enzyme/receptor, toxic targets and ‘silent’ binding sites. Drug molecules distribute in the body according to the rules of thermodynamic distribution processes. When no active transport or permeability barrier exists, the free concentration of the drug molecule should be equal throughout the body, i.e. in plasma, tissues and other bio-phases. It is desirable to administer the smallest possible dose of the drug that achieves the highest possible free concentration at the site of action with minimal binding to nonspecific or toxic binding sites. Recently, Braggio et al8 introduced the concept of drug efficiency relating the in vivo bio-phase concentration to the dose. Candidate molecules with high in vivo drug efficiency should exhibit good efficacy at lower dose levels, minimising compound availability to bind sites other than the target. Due to the high prevalence of phospholipids and proteins (mostly albumin), even weak binding of the drug molecules significantly reduces the free concentration of the drug at the site of action. Bio-mimetic chromatography provides fast, reliable measurements of compound affinity to phospholipids and proteins. The availability of this information early in the drug discovery process is crucial for effective prioritisation of compounds for further progression and in vivo experimentation.

Figure 1 Distribution of drugs in vivo depends on various equilibrium and rate processes between compartments
Similarity between drug distribution in vivo and bio-mimetic chromatographic distribution
Perhaps surprisingly, the chromatographic process shows great similarity to the process involved in a compound’s distribution in the body (Figure 2). Introduction of a compound into a chromatographic system consisting of a flowing aqueous mobile phase and a lipophilic bio-mimetic stationary phase results in the compound interacting with the stationary phase and being retained, effectively losing speed relative to the mobile phase. Similarly, after oral or intravenous administration, drug molecules go into the continuously flowing bloodstream and interact with the stationary cell and tissue components, and as a result, they are retained in the body.

Figure 2 Illustration of the chromatographic process. Molecule 2 has a greater affinity for the stationary phase and consequently is retained more than molecule 1. As a result, molecule 1 has a lower retention time and is eluted from the column first
Both the chromatographic and biological interactions are mostly reversible and obey the rules of thermodynamic equilibrium processes. In both cases, the compound concentration is much smaller than the available sites of interactions. Drug distribution in vivo occurs on the surfaces of large bio-molecules, resulting in a dynamic equilibrium distribution, as in chromatography. These interactions and dynamic equilibrium processes are more entropy driven than the octanol/water partition process that is often used to model compounds’ biological distribution9. In bio-mimetic chromatography, a compound is injected into a moving aqueous mobile phase and the compound interacts with the stationary phase – typically, chemically bonded large biomolecules such as human serum albumin (HSA), phosphatidyl choline (IAM), etc. Compounds with a high affinity for the stationary phase spend more time associated with that phase and are retained longer on the column. When the compound is eluted from the columns, it appears as a peak on the chromatogram and the peak maxima is measured as retention time. The retention time (tR) expressed relative to the retention of unbound solvent molecules (t0) is directly proportional to the compounds’ average distribution between the mobile and stationary phases (see Equation 1).

Equation 1
The chromatographic process is also highly selective, as it is normally used for the separation of compounds often closely related in structure. Optically active stationary phases such as proteins are able to separate optical isomers. There have been several examples in the past when the optical isomers of a drug molecule exert very different pharmacokinetic behaviour10.
Importance of plasma protein binding
Pharmacokinetic parameters are usually derived from total plasma concentration of a drug without taking into account whether the drug is free or bound to plasma proteins. Recently, free drug concentration in plasma has received greater attention as it often shows better correlations with drug efficacy than the total plasma concentration11. The explanation for this is that the free concentration of the drug in plasma is related to the free concentration of drug available at the site of action7. Although plasma contains many components, the most important proteins that are capable of binding drugs specifically (and non-specifically) are human serum albumin (HSA) and alpha-1-acid glycoprotein (AGP). Typically, plasma proteins contain 60 per cent HSA and one to three per cent AGP, the remainder consists mainly of neutral immuno-globulins. The physiological role of HSA is to transport fatty acids and bind other potentially toxic compounds that need to be cleared from the body. The exact physiological role of AGP is not known, but its physiological concentration may increase significantly in certain disease states, such as inflammation12, and it varies with age, gender and ethnic origin13. Therefore, drugs which bind strongly to AGP may display variable efficacy and pharmacokinetics. Traditional methods for determining plasma protein binding include ultra-filtration and equilibrium dialysis, both of which are time consuming with limited capacity and require expensive analytical instrumentations (HPLC with mass spectrometric detection). For both methods, accurate quantification for strongly bound compounds is severely hampered by the detection sensitivity limits. Therefore, none of these techniques are suitable for confidently ranking compounds that bind above 99 per cent. Conversely, the chromatographic approach does not suffer such limitations and can effectively differentiate between extremely high binding compounds.
Chromatographic measurements of compounds interactions with albumin The human serum albumin stationary phase was originally developed for optical isomer separations. However, excellent correlations have been reported between the retention times on a HSA stationary phase and the plasma protein binding of known drug molecules14,15. For such correlations to be obtained, it is essential that the albumin stationary phase contains the protein in as close to its native conformation as possible. To facilitate this, the mobile phase pH should be kept at the physiological plasma pH (pH=7.4). The crystal structure of human serum albumin has revealed various binding sites, such as the so-called warfarin binding site, benzodiazepine binding sites and six sites that can bind fatty acids16. The integrity of these binding sites (and of the protein as a whole) may be evaluated by studying the performance of suitable standard compounds, such as warfarin in the bio-mimetic chromatographic system.
The importance of compound affinity for specific binding sites in relation to drug-drug interaction is well known, when two different drug molecules compete for the same binding site and may displace each other, thus altering the free concentration. This may result in an increase above the required therapeutic level and even reach a dangerously high toxic level17. Many compounds have only weak, non-specific binding to the albumin, which is often ignored. This non-specific binding often relates to the hydrophobicity of the compound. An analysis of albumin binding data for over 100,000 research compounds (including known drug molecules) revealed that lipophilicity/hydrophobicity is a major factor in this non specific binding. Additionally, the presence of negative charge creates strong interactions with the cationic region of the albumin’s major binding site; aromatic rings facilitate π-π interactions, further increasing the binding. The presence of a large hydrophobic pocket close to the positively charged surface of the albumin also modulates the binding strength of molecules that have hydrophobic substituents close to the negative charge18. Thus, it is possible to reveal structure – binding relationships using the chromatographically determined albumin binding for early drug discovery compounds. This information is very useful for chemists to design molecules with more advantageous binding characteristics. An example from an in-house drug discovery program is shown in Figure 3. We have observed that moving the ether oxygen in the aliphatic substitution reduces the binding regardless of other structural modifications. These compounds all had a carboxyl functionality, which was required for potency but unfortunately caused strong specific binding of the molecules to albumin. However, making the alkyl side chain more polar, especially at the end by introducing the ether oxygen, reduced the hydrophobic interactions.

Figure 3 General and specific structure – albumin binding relationships for an in-house research program
Using the Abraham solvation equation model, it has been shown that the non-specific albumin binding is similar to the octanol/water partitioning. In both cases, the H-bond acidity of the compound does not influence the binding and partition19,20. It is an important finding as the octanol/water partition coefficient and it’s calculated in silico version (clogP/clogD) is often used to model a compound’s in vivo distribution and non-specific binding. However, in the presence of an acidic moiety, there is a significant difference between HSA binding and octanol/water partitioning. When an acidic group is present on the molecule, the octanol/water distribution coefficient will be lower at pH 7.4 due to the ionisation. Conversely, the albumin binding gets stronger, thus potentially the octanol/water logD values can mislead the medicinal chemist regarding the albumin binding.
Chromatographic measurements of compounds binding to Alpha-1-acid glycoprotein (AGP)
The exact physiological role of the alpha-1-acid glycoprotein in plasma is not known. AGP is predominantly synthesised in hepatocytes and distributed in various body fluids. AGP is a negatively charged acidic glycoprotein that consists of a single polypeptide strand and five carbohydrate moieties. The total carbohydrate content reaches 41 per cent, and the negatively charged sialic acid content reaches up to 11 per cent (Figure 4). The AGP HPLC stationary phase was originally developed for chiral separation purposes. However, the AGP stationary phase, when used with pH 7.4 aqueous mobile phase and a shallow gradient only up to 25 per cent iso-propanol, can be utilised to study the binding of compounds under physiological conditions23. The gradient HPLC retention time obtained on the AGP column shows excellent correlation with the AGP binding data obtained by ultra-filtration methods23. The structure – binding relationship studies revealed that AGP binds positively charged hydrophobic molecules very strongly. AGP binding does not show any correlation to albumin binding.

Figure 4 The ribbon molecular model of AGP in the native state and its hypothetical binding sites21,22
We have developed a model for the estimation of the total plasma concentration of research compounds using the chromatographically determined HSA and AGP binding data. The size of the molecules or the chromatographically determined lipophilicity24 had to be included in the model to account for a compound’s non-specific binding to neutral immuno-globulins. Strong/specific AGP binding of compounds can be identified by comparing the binding strength with a compound’s lipophilicity. In several cases, this data helped to explain the differences of the in vitro and in vivo clearance of project compounds. For example, for a specific project, the compounds showed high in vitro clearance as opposed to slow in vivo clearance. When AGP was added to the in vitro experiments, the in vitro clearance showed the same level as it was observed in vivo.
Chromatographic measurements of compounds binding to phospholipids
Beside the plasma proteins, there is another very important constituent of the body, namely the phospholipids. Phospholipids can be found in every cell and some tissues contain large proportions. There are several types of phospholipids, such as neutral (phospho inositides), acidic (phosphatidic acid), basic (phosphatidylethanolamine) and zwitterionic, like phospathidylchloline. The most abundant is phosphatidylcholine, the main component of lecitine. Pidgeon and co-workers25 developed the Immobilised Artificial Membrane (IAM) HPLC stationary phases in an attempt to simulate the lipid environment in a cell on solid surfaces. The IAM stationary phase contains phosphatidyl choline chemically bonded to silica as shown in Figure 5. Compound retention time on the IAM stationary phase can be related to a compound’s binding to phospholipids. It has been shown that the chromatographic retention on IAM exhibits good correlations to oral absorption26, brain tissue binding27, and other biological disposition28. We have reported29 that a compound’s binding to IAM shows a correlation with the compound’s lipophilicity. An Abraham solvation equation analysis generated on a standard set of diverse compounds revealed that H-bond acidity had again a zero coefficient, similarly to the octanol/water partition29. However, positively charged compounds showed very strong IAM binding, especially with elliptical molecules such as para-substituted phenyl rings and with piperazine, for example. In this respect, the binding is similar to the AGP binding. However, the Abraham solvation equation generated for the description of AGP binding revealed a significant difference. The H-bond acidity parameter had a large negative coefficient which means that H-bond donor compounds have reduced binding to AGP, relative to IAM binding or octanol/water partition (unpublished results).

Figure 5 The chemical structure of IAM column and the phyosphatidylcholine bi-layer of the biological membranes
Models to predict in vivo volume of distribution and drug efficiency
The above mentioned bio-mimetic chromatographic methods have been published6,14,15,18,22,26,28 and used to reveal structure-binding relationships. However, there is a need for clear guidelines to efficiently utilise these data, ensuring that excessive ‘off target’ affinity does not prevent compound progression. In-depth analysis of these data has yielded great insight into the in vivo behaviour of compounds and enabled the creation of new tools of great utility early in the drug discovery process. It is well accepted that drugs that require a very low dose with the least frequent administration are preferable. Early knowledge of the required dose for a research molecule to achieve clinical efficacy without toxic side effects would be beneficial in compound selection and progression. A principal goal is to maximise the in vitro potency of the molecules. It is well known that compounds displaying similar in vitro potencies might require very different dosing regimes for clinical efficacy because of different in vivo distributions. These differences are caused by the different absorption and pharmacokinetic properties of the compounds.
For example, the steady state volume of distribution is an important in vivo parameter and provides an indication of the distribution of the drug between the plasma and tissue compartments. The in vivo steady state volume of distribution is defined as the dose divided by the plasma concentration. It is usually derived from the plasma concentration – time profile30. It has been long recognised that positively charged lipophilic compounds have high volumes of distribution, while acids have very low volumes of distribution31. This observation suggests that compounds that bind more strongly to albumin have low volumes of distribution while compounds binding strongly to IAM have high volumes of distribution. When we applied this hypothesis to the data for over a hundred known drug molecules, a statistically significant model was obtained32. It is not surprising as the plasma compartment contains albumins and other proteins but does not contain phospholipids, whilst tissues contain both phospholipids and albumin type proteins. Therefore, the enhanced phospholipid binding will drive the compound towards the tissue compartment (see Figure 6). Other models published in the literature using in vitro, physico-chemical data yield similar results. The Pfizer model33 for volume of distribution uses ElogD to model lipophilicity, fraction of positive charge and plasma protein binding, while the Bayer model34 estimates the volume of distribution by applying the measured phospholipid and HSA binding data obtained on immobilised phosphatidylcholine and serum albumin on silica surface.

Figure 6 Relationship between IAM and HAS binding of known drug molecules and the influence of charge (blue: positively charged at pH 7.4; red: negatively charged at pH 7.4; green: neutral at pH 7.4) and schematic representation of volume of distribution and contributing factors with the model equation
While the volume of distribution reveals the distribution of drug between the plasma and tissue compartment, it is even more important to know how the compound distributes as free and bound in the body enabling the calculation of the required dose for a certain free concentration at the site of action. When the free drug hypothesis is valid (no significant active transport between compartments and no permeability restriction), the free concentration in the plasma gives an indication of the free concentration in the bio-phase. The unbound volume of distribution (Vdu) is obtained from the proportion of the dose and free plasma concentration. The reciprocal value of the unbound volume of distribution can be considered as the maximum drug efficiency of the compound (DRUG eff max) which indicates the maximum bio-phase concentration at the target when 100 per cent absorption, no active transport and no permeability restriction occur.
The unbound volume of distribution can be calculated from the volume of distribution and plasma protein binding, usually expressed as fraction unbound (fu); specifically Vdu = Vd/fu. The unbound volume of distribution in principle tells us what will be the steady state free plasma concentration at a given dose (see Figure 7). When we know the potency data of a research molecule (usually available in early drug discovery from high throughput screening as pIC50 or pKi) and know the drug efficiency of the compound, we can estimate the required dose to obtain 50 per cent inhibition/receptor occupancy of the compound in vivo. In this way, we can take into account not only the potency but also the in vivo availability (efficiency) of the compound at a given dose (we suppose 100 per cent absorption or intravenous administration).

Figure 7 The relationships between free plasma concentration, dose and potency
We have found that the unbound volume of distribution and thus the in vivo DRUG eff max can be estimated from the proportioned sum of the IAM and HSA binding data obtained from the fast gradient bio-mimetic HPLC measurements35 (see Figure 8). We have used human clinical volume of distribution and plasma protein binding data for more than 100 known drug molecules for constructing the model equation.

Figure 8 The agreement between the in vivo unbound volume of distribution and the estimated unbound volume of distribution from the HPLC based IAM and HSA binding
These models make a reasonably good estimation of a compound’s in vivo distribution possible and the availability at the site of action by simply measuring their albumin and phospholipid binding using bio-mimetic HPLC.
Estimation of tissue binding
The volume of distribution reveals how a drug molecule distributes between the plasma and tissue compartment, but it does not provide information on how the compound distributes between various tissues. For drugs that are designed to go to the brain for the required pharmacological effect, we would prefer high brain tissue accumulation together with high free brain concentration. For drugs designed to cure lung diseases for example, we would prefer that the compounds partition preferentially into the lung. Different tissues contain various amounts and types of proteins and phospholipids. Therefore, we can assume that an appropriate combination of the bio-mimetic binding properties show good correlation with various tissue binding data. Tissue binding can be measured by using the equilibrium dialysis tissue homogenates with spiked drug molecules36.
As an example, we have found good agreement between lung tissue binding and bio-mimetic binding data (manuscript is in preparation) on known drug molecules. Lung tissue contains a high proportion of mucus, which contains mucin, which is mainly highly glycosilated protein, just like AGP. Therefore, it is not surprising that the AGP binding plays a dominant role in lung tissue binding, as shown by the high coefficient of the AGP binding in the model.
For an estimation of brain tissue binding, the dominant factors are the HSA and IAM binding, highlighting the difference in composition of brain tissue from lung tissue (in-house model).
Applications in lead optimisation – strategies
Discovering potent molecules and understanding the structure – binding relationships with the drug target still remains the primary driver for compound selection. Taking into account crystal structures and using the available structure based design tools helps to synthesise thousands of potent molecules. When the compounds with good potency have been identified, we need to take into account ligand efficiency37,38, that relates the potency to the size, polar surface area or the lipophilicity of the compounds. The next step is to move our attention to the in vivo pharmacokinetic/ pharmaco-dynamic aspects when selecting potent compounds for progression (see Figure 9 ).

Figure 9We have to balance the potency and the drug efficiency of the molecules at early stages of drug discovery (Green: known drug molecules; Purple: GSK research program compounds
We have found that the drug efficiency and the ligand efficiency often showed good correlation as both require low lipophilicity and high specificity for binding mainly to the target and not the albumins and phospholipids. We have also observed that the solubility of the efficient molecules is usually higher than the less efficient ones. In general, the drug efficiency increases with decreasing lipophilicity as shown for 137 known drug molecules in Figure 10. Consequently, we recommend that bio-mimetic HPLC measurements of albumin and phospholipid binding are made soon after the potency measurements to allow them to be used in combination with the potency for prioritisation of the compounds for in vivo measurements.

Figure 10 Inverse correlation of Drug efficiency and logP
In conclusion, high throughput chromatographic measurements of compounds’ binding to proteins and phospholipids can be invaluable in lead optimisation and compound pro – gression. Experimentally measured bio-mimetic binding data have been used to build models for the estimation of compounds’ in vivo distribution. It has been found that although receptor binding, protein binding and phospholipid binding are all governed by a compound’s lipophilicity, the simple octanol/water partition coefficients are not able to describe the different distribution of acids and bases in vivo. The bio-mimetic chromatography has proved to be a very good model to describe the in vivo non-specific binding of drugs.
References
1. Van de Waterbeemd, H. and Gifford, E., ADMET in silico modelling: towards prediction paradise? Nature Rev Drug Discov, 2003.2: p.192-204
2. Chohan, K.K., Paine, S. W., Waters, N. J., Advancement in predictive in silico models for ADME. Curr Chem Biol, 2008. 2 (3): p.215-28
3. Stouch, T. R., Kenyon J.R., Johnson S. R., Chen X-Q., Doweyko, A., Li, Y., In silico ADME/Tox: why models fail. J Comp-Aided Mol Design, 2003. 17: p.83-92
4. Feng, M. R., Lou, X., Brown, R.R., Hutchaleelaha, A., Allometric pharmacokinetic scaling: towards the prediction of human oral pharmacokinetics. Pharm Res, 200. 17(4): p.410-18
5. Mahmood, I., Theoretical versus empirical allometry: Facts behind theories and application to pharmacokinetics. J Pharm Sci, 2010. 99 (7):p.2927-33
6. Valko, K. Application of high performance liquid chromatography based measurements of lipophilicity to model biological distribution. J Chrom A, 2004. 1037:p299-310
7. Brodie, B. B., Kutz, H., Schanker, L. J., The importance of dissociation constants in lipid solubility on influencing the passage of drugs into the CSF. J Pharmacol Exp Ther, 1960. 130:p.20-25
8. Braggio, S., Montanari, D., Rossi, T., Ratti E., Drug efficiency: a new concept to guide lead optimization program towards the selection of better clinical candidates. Expert Opin Drug Discov, 2010. 5(7): p.609-18
9. Dorsey, J. G., Khaledi, M. G., Hydrophobicity estimations by reversed-phase liquid chromatography; Implications for biological partitioning processes. J Chrom A, 1993. 656:p.485-99
10. Levy R. H., Boddy, A. V., Stereoselectivity in pharmacokinetics: A general theory. Pharm Res, 1991. 8 (5): p.551-55
11. Trainor G. L., The importance of plasma protein binding in drug discovery. Expert Opin Drug Discov, 2007. 2(1):p. 51-64
12. Israili, Z. H., Dayton, P. G., Human alpha-1- glycoprotein and its interactions with drugs. Drug Metab Rev, 2001. 33(2):p.161-235
13. Johnson, J. A., Livingston, T. N., Differences between black and whites in plasma protein binding of drugs. Eur J Clin Pharmacol, 1997. 51:p.485-88
14. Noctor, T. A. G., Diaz-Perez, M. J., Wainer I. W., Use of human serum albumin-based stationary phase for high-performance liquid chromatography as a tool for rapid determination of drug plasma protein binding. J Pharm Sci, 1993. 82:p.675-76
15. Tiller, P.R., Mutton, I. M., Lane, S. J, Bevan, C.D., Immobilised human serum albumin: Liquid chromatography/mass spectrometry as a method of determining drug-protein binding. Rap Comm Mass Spectrom, 1995. 9:p.261-63
16. Bhattacharya, A. A., Grune, T., Curry, S., Franks, N. P., Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J Mol Biol 2000. 303:p.721-32
17. Tesseromatis, C., Alevizou, A., The role of the protein-binding on the mode of drug action as well the interactions with other drugs. 2008. 33(4):225-30
18. Noctor, T. A. G., Pham., C. D., Kaliszan, R., Wainer, I. W., Stereochemical aspects of benzodiazepine binding to human serum albumin. Enantioselective high performance liquid affinity chromatographic examination of chiral and achiral binding interactions between 1,4- benzodiazepines and human serum albumin. Mol Pharmacol 1992. 42(3):p.506-11
19. Valko, K. Nunhuck, S. Bevan, C., Abraham, M. H., Reynolds, D. P., Fast gradient HPLC method to determine Compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J Pharm Sci, 2003. 92(11): p.2236-48
20. Abraham, M. H., Scales of solute hydrogenbonding: their construction and application to physicochemical and biochemical processes. Chem Soc Rev, 1993. 22:p.73-83
21. Kopecky V. Jr., Ettrich, R., Hofbauerov, K., Baumruk, V., Structure of human alpha-a-acid glycoprotein and its high affinity binding site. Biochem Biophys Res Comm 2003. 300(1): p.41-6
22. Kaliszan, R., Nasal, A., Turowski, M., Binding site for basic drugs on α1- acidglycoprotein as revealed by chemometric analysis of biochromatographic data. Biomed Chrom 1995. 9:p.211-15
23. Rapti A., Evaluation of alpha-1-acid glycoprotein binding measurements by high performance liquid chromatography and ultrafiltration method. Drug Discov MSci student’s report, University of London, School of Pharmacy, 2006. p.53-63
24. Valko, K., Du, C.M., Bevan, C., Reynolds, D. P., Abraham, M. H., Rapid method for the estimation of octanol/water partition coefficient (logPoct) from gradient RP-HPLC retention and a hydrogen bond acidity term (Σα2H). Curr Med Chem 2001. 8:p.1137-46
25. Pidgeon, C., Venkataram, U.V., Immobilised artificial membrane chromatography: supports composed of membrane lipids. Anal Biochem 1989. 176:p.36-47
26. Stewart, B., Chan, H., Use of Immobilized artificial membrane chromatography for drug transport applications. J Pharm Sci 1998. 87:p.1471-78
27. Salminen, T.m Pulli, A., Taskinen, J., Relationship between Immobilised artificial membrane chromatographic retention and the brain penetration of structurally diverse drugs. J Pharm Biomed Anal 1997. 15:p.469-77
28. Lazaro, E., Rafols, C., Abraham, M. H., Roses, M., Chromatographic estimation of drug disposition properties by means of immobilised artificial membranes (IAM) and C18 columns. J Med Chem 2006. 49:p.4861-70
29. Valko, K., Du, C. M., Bevan, C. D., Reynolds. D. P., Abraham, M. H., Rapid gradient HPLC method for measuring drug interactions with immobilised artificial membrane: Comparison with other lipophilicity measures. J Pharm Sci 2000. 89 (8):p.1085-96
30. Kwon, Y. Handbook of Essential Pharmaco – kinetics, Pharmacodynamics and Drug Metabolism for Industrial Scientists. Springer-Verlag. 2001. New York p. 73-82
31. Smith, DA, in Pharmacokinetics and metabolism in drug design. Smith, D.A, van de Waterbeemd, H, Walker, D. K., Mannhold, R., Kubinyi, H., Timmerman H. Eds. Wiley, New York, 2001. Pp.51-55
32. Hollosy, F., Valko, K., Hersey, A., Keri, Gy., Bevan, C., Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. J Med Chem 2006. 49:p.6958-71
33. Lombardo, F., Obach, R. R., Shalaeva, M. Y., Gao, F., Prediction of volume of distribution values in humans for neutral and basic drugs using physicochemical measurements and plasma protein binding data. J Med Chem 2002. 45:p.2867-76
34. Willmann, S., Lippert,J., Severstre, M., Solodenk, J., Schmidt, W., PK-SIM: a physiologically based pharmacokinetic “whole-body” model. Biosilico 2003. 1:p.121-24
35. Valko, K., Nunhuck, B., Hill, A.P., Estimating unbound volume of distribution and tissue binding by in vitro HPLC based Human Serum Albumin (HSA), and Immobilised Artificial Membrane (IAM) binding measurements. J Pharm Sci 2010. 10: (in press)
36. Wan, H., Rehngren, M., Giordanetto, F., Bergstrom, F., Tunek, A. High-throughput screening of drug-brain tissue binding and in silico prediction for assessment of central nervous systems drug delivery. J Med Chem 2007. 50:p.4606015
37. Abad-Zapatero, C., Metz, J. T., Ligand efficiency indices as guideposts for drug discovery. Drug Disc Today 2005. 10(70):464-9
38. Leeson, P. D., Springthorpe, B., The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 2007. 6(11):p.881-90
About the Author
Klara Valko
Klara graduated as a pharmacist and received her PhD from School of Pharmacy, Semmelweis University in Budapest, Hungary in 1979. She has published over 90 peer-reviewed papers in the field of structure – chromatographic retention relationship and its application in drug design. She was awarded the Doctor of Science degree by the Hungarian Academy of Sciences in 1996. She has been working in the Physico-Chemical Characterisation Group at GlaxoWellcome and now GSK since 1995. She has developed methodology and models for the application of bio-mimetic HPLC measurements for the estimation of in vivo distribution of drugs. She has applied the methodology to support over 20 neuroscience programmes within GSK. In addition to the work at GSK, she was appointed visiting professor at London School of Pharmacy, where she has been teaching the Physchem/ADME Module to the Drug Discovery MSci students since 2002.





