Quality by Design for generic products: opportunities and challenges
Posted: 13 December 2016 | Patrick Crowley, Sean McCrossen | No comments yet
Quality by Design (QbD) precepts provide opportunities for enhancing efficiencies and reducing costs, especially for generic products. Over 80% of prescriptions dispensed in the United States in 2012 utilised generic dosage forms and it is unlikely that this proportion will increase to any extent because fewer innovative drugs are set to lose patent protection.

Consequently, Generics organisations must compete in terms of procurement, manufacture and testing efficiencies. QbD concepts, if embraced, explored and implemented can help gain competitive edge. Perspectives and suggestions on how generics manufacturers might avail of such opportunities are proposed and discussed in this two-part article, focusing on dosage form design and manufacture, monitoring and control. Part 2 will be published in Issue 1, 2017 of European Pharmaceutical Review.
QbD definitions and terminologies are not discussed in these articles. These are readily sourced from the many guidelines and publications on the topic. Neither is this a comprehensive review of the state of the art. Rather, the intent is to suggest some possibilities for QbD-related efficiencies that may be readily applied to new and existing medications. The focus is largely on solid oral dosage forms, these being the most prominent medicinal products.
The promise of QbD
QbD precepts are ostensibly simple, viz: a product and the process for its manufacture is developed to meet pre-defined performance and quality objectives. Relationships between product composition, attributes of the input materials, the process for product manufacture and the attributes of the dosage form are well understood. Such information could conceivably deliver the following efficiencies, in addition to providing patients with more consistent and better quality medications:
- Sourcing flexibility with respect to input active pharmaceutical ingredient (API) and excipients
- Flexibility with respect to equipment type, processing parameters, site and scale of manufacture
- More efficient operations based on ‘real time’ (continuous) quality or performance-monitoring rather than time-based endpoints
- Replacement of much end-product testing with ‘at line’ or online monitoring
- Reduced stability testing workloads
- Faster manufacture, testing and batch review/approval times
- Fewer resources required in regulatory, quality and testing groups.
The benefits for manufacturing efficiencies, supply chain flexibility and cost of goods are self-evident.
QbD can be applied to existing as well as new products provided that appropriate product and process knowledge is generated, validated and implemented. Comprehensive knowledge for effective implementation of all possible QbD-related efficiencies may not be available at filing, however, regulatory agencies now espouse and welcome a ‘continuous improvement’ ethos rather than the previous ‘three batches then lock down the process’ viewpoint.
The United States Food and Drug Administration has also stated that organisations should ‘‘be bold’’ in their thinking when registering products and processes that have been developed using a QbD approach. Good science must always underpin programmes to enhance efficiencies and economies of manufacture and supply. Such science must be clearly expounded in regulatory filings and be available to guide aid Agency inspections at manufacturing plants.
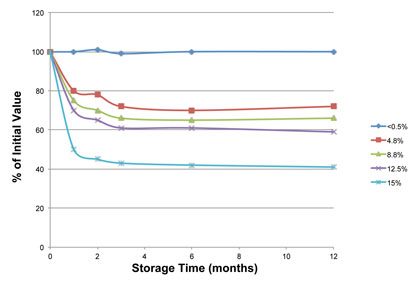
Figure 1: Stability of ‘Compound A’
Dosage form design
A generic product seeks to replicate the performance of the innovator drug with respect to amount and quality of the active ingredient, clinical efficacy and overall quality. Such a target quality product profile (TQPP) is defined as “a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the drug product” (ICH Q8). In Table 1 we suggest a suitable TQPP for an orally administered generic product so that the product is designed to be fit for purpose to treat clinical conditions safely and effectively.
Table 1: Target quality product profile for a generic product
| Drug content identical to that in innovator product |
| Biopharmaceutically equivalent to innovator product |
| Product quality of drug is comparable to that of innovator product throughout product life |
| Consistent, reliable and cost-effective manufacture |
| Viable shelf life and comparable storage requirements to innovator |
Such targets can generally be defined at the outset with generic products and do not usually change.
The requirements in Table 1 for drug content and bioequivalence ensure that the product is therapeutically equivalent to the innovator product. Drug quality, in terms of purity, residues, structural deviants (impurities) and degradation products must also be at least as good as in the innovator product. Additional requirements might concern appearance, shape and other patientcentric attributes. For example a smaller oral dosage unit could be advantageous and be facilitated by the advent of novel manufacturing technologies or excipients that were not available when the innovator product was developed.
Table 1 does not stipulate that drug dissolution rate be comparable to that of the innovator product because such comparability does not guarantee bioequivalence. Regulatory agencies have, for some time, encouraged the development of in vitro-in vivo correlation (IVIVC). However, despite many decades of efforts, examples of such association are rare: the gastrointestinal tract is too variable. Even if an association had been developed by the innovator this would likely be formulation and component (excipient)- specific. Sugita et al1 showed that excipients could affect the particle size of precipitated pioglitazone in the gastrointestinal tract and reduce bioavailability, despite comparable in vitro dissolution rate.
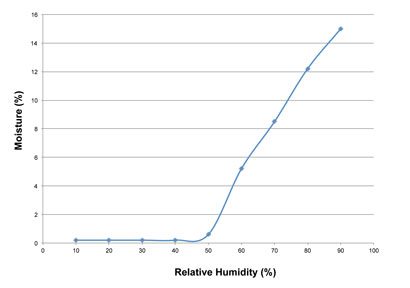
Figure 2: Equilibrium moisture contents at different relative humidity values
Unless the generic organisation can exactly replicate the composition and process for the now-mature innovator product there is a risk of bio-inequivalence. Such considerations mean that ‘bioequivalence’ rather than dissolution rate equivalence is a more appropriate target. Lionberger et al propose that only patientrelevant attributes be listed in the TQPP2 . This may be desirable from a regulatory perspective but may not provide the requisite guidance to groups tasked with designing a product and process for its manufacture. Efficiency and reliability in manufacture and testing are key considerations in the competitive environment of generic medicines. It is important, therefore, that groups tasked with dosage form and process design are provided with the requisite focus and targets at the outset. Process-related requirements are accordingly included in Table 1. The term ‘Product Design Specification’ has also been suggested, as a means of incorporating efficiency targets in a development programme, however, such proliferation of terminologies can confuse the reader and we do not use them here.
Replicating the composition and possibly the process of the innovator product might seem the simplest way to assure comparable performance. This strategy may be short-sighted for a number of reasons:
- It may be relatively easy to identify the excipients in the innovator product but grade or other details (for instance, the chain length of polymerised celluloses) may not be readily established. Product performance requirements might also have changed since the innovator product was first brought to market. For instance, alcohol-induced dose dumping and its impact on patient safety has become a major concern with some modifiedrelease products. Release-modifying materials for a generic product might need to be chosen with more stringent requirements in mind
- The generics company may be knowledge-rich with respect to facilities, equipment and materials. Such in-house expertise and resource could be invaluable. In contrast, the process for manufacture of the innovator product may utilise equipment, materials and processes that have remained unchanged since the product was first made available. There may be opportunities to develop a more efficient process, using different excipients, equipment and processes.
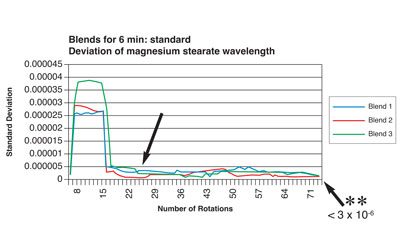
Figure 3: Relationship between number of blender rotations and magnesium stearate dispersion homogeneity by NIR analysis (reproduced with permission from reference)
Knowledge of the API’s properties
Capitalising on efficiency-related opportunities requires knowledge of the properties of the active pharmaceutical ingredient (API) so that attributes critical to product quality and process viability and efficiency are reflected in product and process design. However, there may be little relevant information in the public domain. Compendial monographs provide little more than summary statements on aspects such as purity, impurities, solubility and hygroscopicity. They do not provide detailed information on drug stability or mechanisms/modes of degradation and of other less tangible but important behavioural nuances and vagaries that can affect processing or quality attributes. Such information is usually confidential to the innovator company. A relatively simple example concerns a putative ‘compound A’ that is sensitive to, and extensively degraded by, residual moisture.
Figure 1 shows that compound A is stable at moisture contents at and below 0.5% but is rapidly and progressively degraded at higher moisture contents. Hence, moisture content is a critical material attribute. Figure 2 shows that compound A acquires moisture (is hygroscopic) when exposed to environmental relative humidity (RH) exceeding 50%.
Such attributes have implications for API manufacture, transport and storage. Conditions for drying, isolation and packaging of the API need to be designed to ensure that moisture content remains below the critical 0.5% limit. The RH of environments for drug product manufacture becomes a critical process parameter (CPP). If RH is controlled to a maximum of 50%, compound A does not acquire moisture during processing so is stable. A ‘control strategy’ for drug and drug product to mitigate/eliminate risk might accordingly include:
- Monitoring and controlling environmental RH in processing and packaging facilities
- Formulating with excipients that do not engender RH levels exceeding 50%
- Packaging for API, intermediates and finished product storage that provides a suitable barrier to moisture ingress.
Such understanding, along with monitoring and control of the environmental RH (a CPP), could sustain a case that tests like moisture content on finished product are not relevant nor necessary. Suitable processing and packaging can mitigate the risk of moisture ingress during manufacture and product shelf life: the product remains stable. This would arguably be a better control strategy than one based on ‘end-product’ tests and limits for moisture content, with the attendant risks of false positives due to method variation or acquisition of moisture during sampling or preparation of material for testing.
Not all relationships are so simple but, where these are well researched, developed and given context, a case can be sustained for reduced end product ‘specification testing’.
In the above example product stability studies might require monitoring relative humidity ‘within the pack’ rather than periodic measurement of moisture content, drug content and degradation products and other such conventional tests. Testing programmes could be leaner and rogue results fewer.
Securing a supply chain for product manufacture may require that APIs be sourced from more than one provider. Supplier qualification programmes should confirm the consistency of critical material attributes (CMAs) germane to the specific product and process for multi-sourced materials. Change control agreements should stipulate that prior consultation take place before changes are made by the API provider so that, if necessary, studies are performed to affirm that CMAs are not affected by the proposed change.
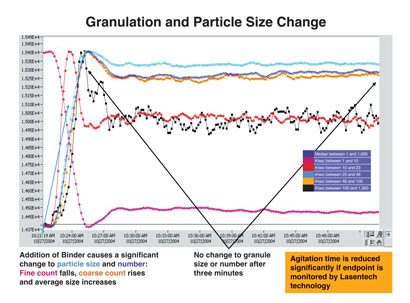
Figure 4: Granulation and particle size distribution (Lasentech) after addition of granulating agent
Excipients
Excipients may also need to be multi-sourced to ensure security of supply. Similar considerations to those pertaining to API sourcing are appropriate. Some excipients may contain low-level (impurities) residues with the potential to interact with the API (see Figure 3, Figure 4 and Table 2). Such residues may not be listed, nor limited in compendial specifications, possibly because levels are low and present no safety risk. However, in cases where the excipient-todrug ratio is high, impurity levels in the excipient may be enough to form significant levels of a novel interaction product, whose safety then needs to be assured (assuming that transfer from excipient to drug occurs during long-term storage). Reformulation using an alternative excipient may be necessary, with attendant programme delays, particularly if such drug-excipient residue reaction rates are slow; degradation products may only reach detectable levels over long-term storage.
Table 2: Potential residues in common excipients*
| Excipient | Residue |
| povidone, crospovidone, polysorbates | peeroxides |
| magnesium stearate, pips | aldehydes, reducing sugar |
| microcrystalline cellulose | lignin, hemicelluloses, water |
| starch | formaldehyde |
| talc | heavy metals |
| dibasic calcium phosphate dihydrate | alkaline residues |
| stearate lubricants | alkaline residues |
| hydroxypropylmethyl/ethyl celluloses | glyoxal |
*reproduced from Reference 4
The presence of residues does not necessarily lead to drugexcipient interaction. The chemistry may not be appropriate with respect to the specific drug. It is also possible that residues may be strongly associated (‘bound’) with the excipient such that desorption and interaction with the drug do not occur; the residue may not be a CMA. Nevertheless, awareness of possibilities and appropriate investigative studies to determine interaction potential represents good risk management and may avoid unwanted surprises during stability testing studies or with commercial product. Unfortunately, providers are generally reluctant to volunteer additional information on excipients, other than assurance of pharmaceutical quality. Assurance concerning compendial status may not suffice for risk management purposes.
This first part of these QbD-related articles has covered how quality is designed in a pharmaceutical product by clearly defining the target quality and performance requirements, for the drug and the associated components. Part Two considers possibilities for manufacturing efficiencies, particularly those concerned with process monitoring and control.
About the authors
 Patrick Crowley is a pharmacist by training and has spent his entire career involved in dosage form design, in the UK and USA. He made significant contributions, as Laboratory Scientist and Department Manager, in the development of 21 novel molecular entities to medicinal products. These included novel chemical entities, fermentation products and biopharmaceuticals. He also participated in and managed groups developing innovative line extensions of commercial products that secured intellectual property rights. Since retiring as a Vice President in Pharmaceutical Development at GlaxoSmithKline he has operated as an independent consultant and lectures as a Visiting Professor at The School of Pharmacy, Kings College, London.
Patrick Crowley is a pharmacist by training and has spent his entire career involved in dosage form design, in the UK and USA. He made significant contributions, as Laboratory Scientist and Department Manager, in the development of 21 novel molecular entities to medicinal products. These included novel chemical entities, fermentation products and biopharmaceuticals. He also participated in and managed groups developing innovative line extensions of commercial products that secured intellectual property rights. Since retiring as a Vice President in Pharmaceutical Development at GlaxoSmithKline he has operated as an independent consultant and lectures as a Visiting Professor at The School of Pharmacy, Kings College, London.
 Sean McCrossen is a chartered chemist and Fellow of the Royal Society of Chemistry. Sean has worked within the pharmaceutical industry for 30 years, specialising in analytical chemistry, chemical development and product support. Sean runs his own consultancy, specialising in CMC technical development and regulatory services to the pharma industry. He currently is working with worldwide pharma clients, regulatory affairs groups and CMO/CROs on both NCEs and generics.
Sean McCrossen is a chartered chemist and Fellow of the Royal Society of Chemistry. Sean has worked within the pharmaceutical industry for 30 years, specialising in analytical chemistry, chemical development and product support. Sean runs his own consultancy, specialising in CMC technical development and regulatory services to the pharma industry. He currently is working with worldwide pharma clients, regulatory affairs groups and CMO/CROs on both NCEs and generics.
References
1. Sugita M, et al. Effect of Excipients on the Particle Size of precipitated Pioglitazone in the Gastrointestinal Tract: Impact on Bioequivalence. AAPS Journal. 2014; 16 (5) 1119-1127 2. Lionberger RA, et al. Quality by Design: Concepts for ANDAs. AAPS Journal. 2008; 10 268-274