Regulatory, clinical and logistics challenges of Advanced Therapy Medicinal Products (ATMPs) in clinical research
Posted: 8 November 2017 | Andrea Zobel, Bridget Heelan | No comments yet
Advanced Therapy Medicinal Products (ATMPs) are new therapeutics that require different procedures for clinical trials compared to traditional medicinal products. It is important to understand the nature and the manufacturing process of an ATMP for set-up and execution of the clinical development and later marketing of these therapies.

ATMPs are biological medicinal products that can be classified into three different types – cell therapy medicinal products (CTMP), gene therapy medicinal products (GTMP) and tissue engineered products (TEP), but they can be also a combination of these classes.1
A CTMP is a biological medicinal product that contains or consists of cells or tissues that have been subject to substantial manipulation so that biological characteristics, physiological functions or structural properties relevant for the intended clinical use have been altered (see Figure 1). They may also consist of cells or tissues that are not intended to be used for the same essential function(s) in the recipient and the donor. CTMPs are administered with the objective of treating, preventing or diagnosing a disease through the pharmacological, immunological or metabolic action of its cells or tissues.2
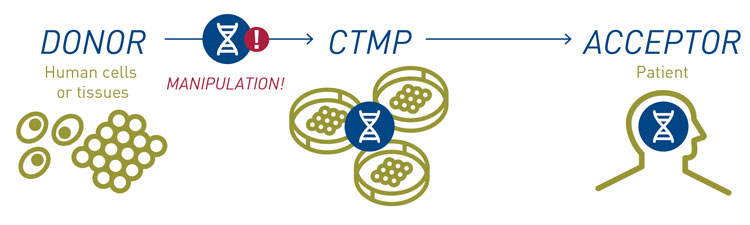
Figure 1: The creation and administration of a CTMP

Figure 2: The creation and administration of a GTMP
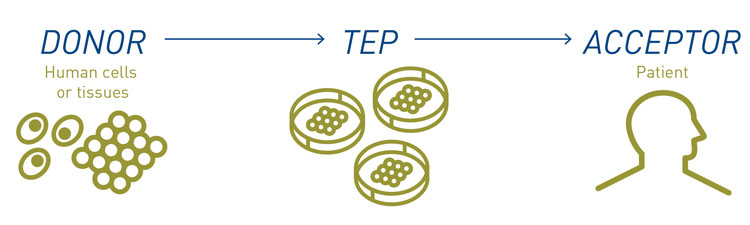
Figure 3: The creation and administration of a TEP
The source of CTMPs can differ: in an autologous transplantation, the CTMP is manufactured from the patient’s own cells or tissue, whereas in allogeneic transplantation, the CTMP is derived from one or a group of healthy donors. An example of autologous transplantation is the chimeric antigen receptor (CAR) T-Cell therapy, where T-cells are isolated from a cancer patient’s blood, genetically modified to express receptors that bind to tumour antigens, and then infused back to the same patient, where these T-cells attack cancer cells that express the targeted antigen on their surface.3 Often, stem cell therapies are allogeneic transplantations, where stem cells are isolated from healthy donors, grown in tissue culture and then infused into patients where they differentiate into tissues or cells to replace defective cells, eg, CNS cells in stroke patients or insulin-producing cells in diabetics.
A GTMP is a biological medicinal product that contains an active substance consisting of a recombinant nucleic acid used in, or administered to humans to regulate, repair, replace, add or delete a genetic sequence (see Figure 2). Its therapeutic, prophylactic or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence.2
Gene therapy can involve the replacement of a missing or defective disease-causing gene. Additional new categories of therapeutic strategies in development include small interfering RNAs.4
Examples of gene therapy targets are the insulin gene in beta cells in diabetes, the sodium channel in lung cells in cystic fibrosis, lipoprotein lipase gene in muscle cells in lipoprotein lipase (LPLD) deficiency.5
TEP (see Figure) refers to a product that contains or consists of engineered cells or tissues, and is presented as having properties for, or is used in or administered to human beings with a view to regenerating, repairing or replacing a human tissue.6
A TEP may contain:
- Cells or tissues of human or animal origin, or both
- Cells or tissues that may be viable or non-viable
- Additional substances, ie, scaffolds or matrices (combined TEP with device).
Regulatory considerations for CTMPs and GTMPs
Table 1: EU guidance and directive for ATMPs
EU Regulation or Directive | Relevant guidance or standards |
ATMP classification | |
Regulation (EC) No 1394/2007 | Provides the definitions of ’tissue-engineering product’ and combined ‘ATMP’ |
Part IV of Annex I to Directive 2001/83/EC | Provides the definitions for ‘gene-therapy medicinal product’ and ‘somatic cell-therapy medicinal product’ |
Genetically modified organisms | |
Directive 2001/18/EC (repealing Council Directive 90/220/EEC) of March 2001 | Puts in place a step-by-step process to assess possible environmental and health risks |
Tissues and cells directive and traceability | |
Directive 2004/23/EC, also known as the European Tissues and Cells Directive | Covers standards for donation, procurement and testing, processing, preservation, storage and distribution of human tissues and cells, as well as its technical implementing directives |
Directive 2006/17/EC, the First Technical Directive | Covers certain technical requirements for the donation, procurement and testing of human tissues and cells |
Directive 2006/86/EC, the Second Technical Directive | Covers standards for traceability, notification of serious adverse reactions and events, and requirements for coding processing, preservation, storage and distribution of human tissues and cells |
Directive 2015/565, amending Directive 2006/86/EC | Certain technical requirements for the coding of human tissues |
Directive 2015/566 | Procedures for verifying the equivalent standards of quality and safety of imported tissues and cells |
The European legal framework provides a lot of guidance and directive for ATMPs (see Table 1). All ATMPs manufactured on a routine basis require a centralised authorisation through the European Medicines Agency. ATMPs are regulated as other biological medicinal products but need more intensive follow-up of efficacy, adverse events and traceability requirements for the market authorisation holder (MAH).
ATMPs, like any other medicinal product, must demonstrate quality, safety and efficacy in order to obtain a licence. However, during development there are additional considerations that need to be taken into account.
For CTMPs or TEPs, one challenge is the donor variability, which requires wide but acceptable product specifications. As many ATMPs have very short shelf-lives (sometimes fewer than three days, and traditional sterility testing takes 14 days) a short shelf-life approach is required. This includes justification of release without full test results and testing of process intermediates, rapid sterility testing and strategies for dealing without specification results post-administration.
Specific methods for characterising the identity and purity of the cell product are required. This could include cell surface markers, gene expression and biological activity, which are used to define the content of the desired cell type as well as product and process-related impurities such as dead cells and carryover of physiologically active components.
For GTMPs, as for any other biological medicinal product, quality should be controlled relative to identity, purity, biological activity and safety. In case of the widely used rAAV (recombinant adeno-associated virus) vector, this can include identity by sequencing of the DNA and Western Blot to test protein expression, purity by residual host DNA/protein, and sterility and biological activity by infectivity (plaque forming units – pfu) and transgene expression. For safety, the sensitivity to detergent and serum free manufacturing to avoid contaminations with, eg, transmissible spongiform encephalopathy (TSE) must be considered.
In general, new development strategies are required; early and continued engagement with regulatory authorities is therefore a prerequisite to success. In general, the regulators are open to discussion on new approaches as long as the approach taken has scientific merit, and does not unduly increase the risk to the patient.
Clinical considerations of ATMPs
Clinical development of ATMPs has specific challenges due to the nature of the medicinal product and the limited availability of non-clinical data. This starts with the challenge of dose finding with often limited PK and PD data. Furthermore, the method of administration can be invasive or require special equipment to place the ATMPs at the right place in the human body, and raises additional procedure-related safety concerns.
The gold standard of a controlled, randomised, double-blind clinical trial may not be feasible or ethically justified for all indications, particularly in life-threatening disease where there is no satisfactory standard of care. For example, to get approval for a trial design which requires a mock surgery is challenging and needs strong justifications. Comparators are often not available and clinical endpoints established for other product types may need adaptation to develop meaningful clinically endpoints reflecting the ATMP activity. Standardisation of the collection and delivery procedure and extended follow-up is required to show efficacy and safety.
Special training is required for all parties to ensure consistency of production, transport and administration. This starts with the collection of starting material, is continued for the staff of the manufacturing and investigational sites, and also includes the clinical team and all involved logistics providers including transportation and import/export providers.
Specific safety issues, such as the risk of integrational mutagenesis of GTMPs, unexpected bio-distribution and ectopic tissue or tumour formation, require a risk management that may need longer follow-up periods, as well as additional trials and registries post-authorisation.
For GTMPs, the correlation between gene expression and pharmacodynamic/functional parameters and bio-distribution of the GTMP needs to be assessed. Vector persistence, oncogenicity, immunogenicity of vector and trans-gene, potential impact of pre-existing immunity, consequences of an immune response on the risks of repeat treatment, viral shedding and risk of third-party transmission should be monitored and controlled in long-term follow up.7,8
For successful global rollout of an ATMP therapy, local regulatory requirements for clinical trials and import restrictions should be considered at an early stage of clinical development. Questions such as whether ATMPs are defined in legislation, whether there are additional regulatory requirements for gene therapy/cell therapy/tissue engineering, or whether there are specific safety/legal restrictions for GMOs and viral vectors, must all be accounted for.
The high levels of process and product characterisation required results in significant costs at all process stages, from establishment of a master cell bank (for allogeneic products), to final product testing. The market conditions and ability of payers to reimburse these very expensive therapies should be checked in advance of the development. When considering the high costs of production, developers must consider the estimated return on investment. Interaction with Health Technology Assessment agencies is advised early in development to ensure that the endpoints chosen in trials will be acceptable not only to regulators but also to payers, in order to ensure reimbursement post-licensing.
Logistics considerations for ATMP trial designs
ATMP trial designs must also be checked for logistics feasibility and processes. Methods and procedures to collect the starting material and to store and supply the finished medicinal products in the supply chain need development in parallel to the clinical development and influence the product significantly.
The supply chain of ATMPs can be divided into four groups: (stem) cell therapy, gene therapy and autologous and allogeneic transplantations (see Figure 4). These include the investigational medicinal product (IMP) logistics and the supply chain of the starting material. The ATMP in a clinical trial is an IMP and the regulations for IMPs must be followed. But the nature of the product creates additional challenges, such as shelf life, storage and shipment conditions, traceability, and labelling and packaging.
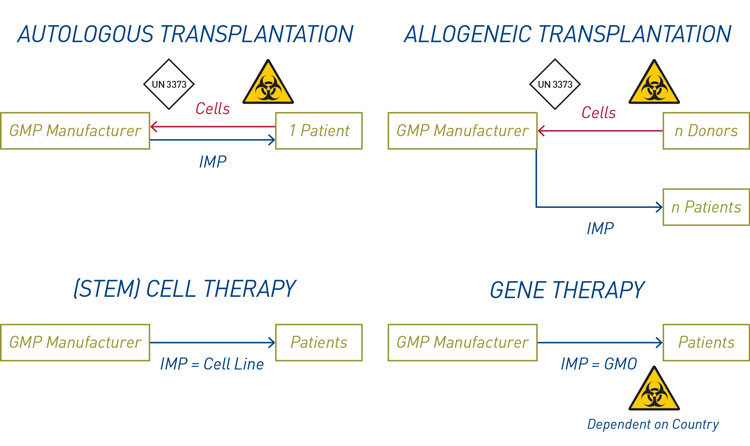
Figure 4: The four groups of the supply chain for ATMPs
Living cells have very short shelf lives of 24 to 96 hours at ambient temperature conditions. This must be considered for the collection of starting material and transportation to the manufacturing site, as well as for the shipment of the finished product to the clinical trial site and administration to the patient. In contrast to traditional clinical studies where clinical sites are selected according to availability of the investigator and access to eligible patients, site choice for ATMP trials must also be based on location in relation to the manufacturing site so that the ATMP can be delivered to patients within its shelf-life. Close cooperation with transportation vendors is required to establish the supply chain.
Shelf-life and temperature are correlated for many biological products. In general, lower temperatures prolong the product’s shelf-life
Cells can be stored long-term in liquid nitrogen (LN2) and shipment containers are available which keep the LN2 for up to two weeks. Shipments in dry ice at -80°C in isolated shipment boxes keep the temperature for some days, but dry ice can also be refilled during transportation. Shipping boxes equipped with phase change material and vacuum panels keep the temperature at refrigerated and ambient temperatures for up to 196 hours (see Figure 5).
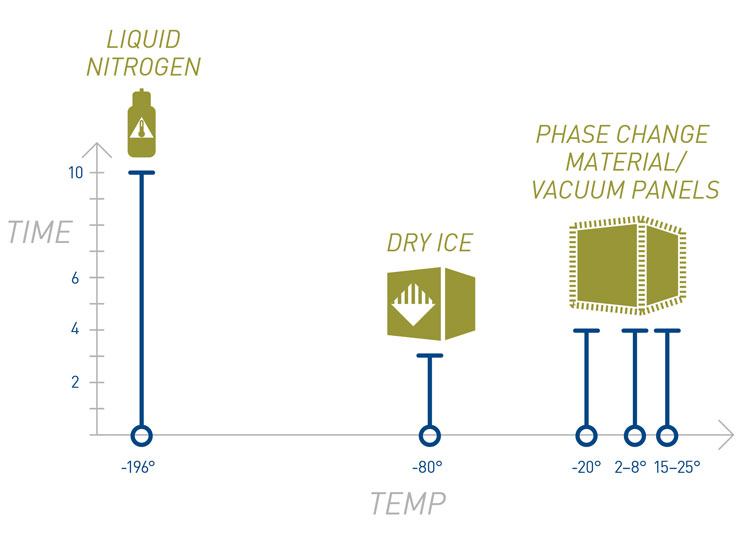
Figure 5: Storage and shipment conditions for ATMPs
Clinical sites must have also the appropriate equipment and licences for storage and preparation of the ATMPs. Deep freezers, liquid nitrogen storage, tissue culture and GMO (genetically modified organism) laboratories are not very common at hospitals and restrict the number of eligible clinical sites. A ready-to-deliver product would ensure quality and consistency between different sites and would be more suitable after licensing. One example is a standardised, clinical and commercial-grade cell therapy product using patented cryopreservation, which allows direct administration without culture at the clinical site.9
Traceability requirements for ATMPs are the same as for IMPs, but require longer archiving, and in the case of cell products, extensive donor documentation including detailed procurement reports of donated materials is required. This is particularly important in the case of autologous transplantations, where the whole supply chain must be documented and traceable at all stages in order to avoid mix-ups and ensure that patients get back their own material. Very sensitive products with short shelf-lives or specific transport conditions could require tracking of location and condition of the shipment through all steps of the supply chain, from collection of the starting material up to delivery to the clinical site. For this purpose, sensors able to measure temperature, humidity shock and light, with real time transmission of data, can be used.
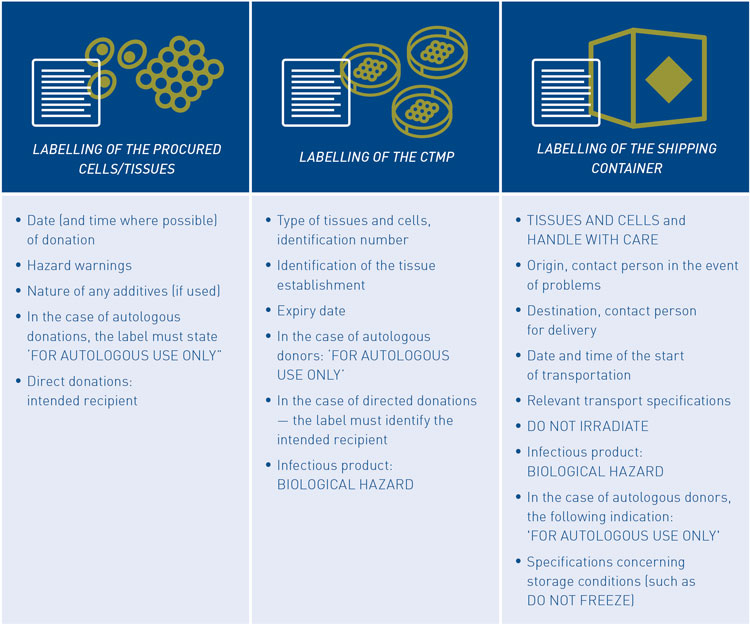
Figure 6: Labelling requirements for ATMPs
Traceability and safety requirements require additional label specifications for ATMP packages and shipping containers (see Figure 6). Warnings to indicate the nature of the product and correct assignment and handling (for autologous use only), should protect the contents of the package from environmental influences (‘Tissues and Cells’, ‘Handle with Care’, ‘Do not Irradiate’, ‘Do not Freeze’), as well as the environment for dangers (‘Biological Hazard’). A dangerous goods classification with the applicable hazard signs could be applicable, for items such as infectious agents and GMOs.
Conclusion
Although only a limited number of ATMPs are currently licensed, scientific and pre-clinical development is advancing in this field and more advanced therapies are entering clinical development. Their different nature in contrast to ‘traditional’ bio-pharmaceuticals influences the regulatory, clinical and logistics processes which are established in clinical research. It is necessary for all involved parties to understand the scientific background and receive the required training to adapt Good Manufacturing Practice and clinical trials to the requirements of ATMPs to ensure that patients receive these products at the correct time, within the shelf-life and that arrangements are in place for long-term follow up for safety and maintenance of efficacy.
About the authors
 Andrea Zobel is Senior Director, Product Management, Clinical Trial Supplies and Logistics, at PAREXEL. Andrea rejoined PAREXEL in 2015 as Global Senior Director, Portfolio Management, and has recently taken on responsibility for the definition and implementation of new products and services for PAREXEL’s clinical trial supplies and logistics business globally. Andrea holds a diploma in Biochemistry from the Freie Universität Berlin and a PhD from the Max-Planck Institute for Molecular Genetics. Contact: [email protected].
Andrea Zobel is Senior Director, Product Management, Clinical Trial Supplies and Logistics, at PAREXEL. Andrea rejoined PAREXEL in 2015 as Global Senior Director, Portfolio Management, and has recently taken on responsibility for the definition and implementation of new products and services for PAREXEL’s clinical trial supplies and logistics business globally. Andrea holds a diploma in Biochemistry from the Freie Universität Berlin and a PhD from the Max-Planck Institute for Molecular Genetics. Contact: [email protected].
 Bridget Heelan is Vice President (Technical), Early Phase, Consulting. Dr. Heelan worked as a consultant immunologist in the NHS before moving to pharmaceutical medicine. At the MHRA she was a senior clinical assessor and Chair of the Rheumatology Immunology Working Party. At PAREXEL she has advised on several clinical development programmes, including ATMPs.
Bridget Heelan is Vice President (Technical), Early Phase, Consulting. Dr. Heelan worked as a consultant immunologist in the NHS before moving to pharmaceutical medicine. At the MHRA she was a senior clinical assessor and Chair of the Rheumatology Immunology Working Party. At PAREXEL she has advised on several clinical development programmes, including ATMPs.
References
- Directive 2001/83/EC, amended by 2003/63/EC annex 1, Part I.
- Directive 2001/83/EC Annex 1, Part IV; amended by 2009/120/EC.
- Jackson HJ, Rafiq S, Brentjens RJ. Nature Reviews Clinical Oncology. 2016;13:370-383.
- Valdmanis PN, Paul N, Kay MA. Human Gene Therapy. 2017;28(4):361-372.
- Gaudet D, Methot J, Dery S, et al. Gene Therapy. 2013;20:361-369.
- EC Regulation No 1934/2007.
- CHMP/GTWP/60436/07
- EMEA/149995/2008
- Sinden JD, Hicks C, Stroemer P, Vishnubhatla I, Corteling R. Stem Cells and Development. May 2017, ahead of print.
Issue
Related topics
Biopharmaceuticals, Drug Development, Gene therapy, Regulation & Legislation