

How to start up an ATMP clinical trial
Posted: 7 July 2020 | Magdalena Czarnecka (KCR) | No comments yet
Advanced therapy medicinal products (ATMPs) are medicines based on genes, tissues or cells. They offer revolutionary opportunities for the treatment of disease and injury; however, developers face a complex regulatory and developmental landscape to bring ATMPs to market. Here Magdalena Czarnecka offers guidance and advice on effectively initiating an ATMP clinical trial.

Situation: what is happening in the ATMP space and why is it of interest?
Research and development on ATMPs has been gathering global momentum since its onset in the late 1980s (Figure 1). Each decade brings an increasing number of ATMP trials, with marketing authorisations first granted in the last 10 years by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). These products are perceived as the third wave of disease management solutions, alongside other widely utilised options based on chemical and biological medicinal products. ATMPs are predominantly investigated in the areas of metabolic diseases, monogenic disorders and oncologic indications with a goal of prevention or treatment and have revolutionary potential, as they carry the promise of potential curative outcomes in addition to long-lasting therapeutic effects.

Figure 1: ATMP Classification (Regulation EC no. 1394/2007)
Complications: what challenges do companies face in delivering these therapies to the market?
Despite the excitement surrounding ATMPs, developers face a complex regulatory and developmental landscape to bring them to market and ultimately to patients. Recent results of a survey-based cohort study1 conducted among commercial European ATMP developers indicate three dominant challenges: regulatory (34 percent), technical (eg, manufacturing, quality standards; 30 percent) and financial (10 percent).
…developers of ATMPs need to confront both challenges related to the clinical trial itself as well as hurdles triggered by the classifications of the investigational medicinal product”
Regulatory challenges are dominated by two issues: 1) the heterogeneity of national procedures and 2) requirements regarding regulatory dossier. Each concept is complex, as the clinical research with these products needs to be compliant both with clinical trial and ATMP-related regulatory requirements (Figure 2). The heterogeneity of national procedures will remain pervasive in the European Union (EU) until the EU Clinical Trials Regulation No 536/2016, harmonising the clinical trial authorisation process, comes into application. The investigational cell therapy or gene therapy medicinal products (CTMP or GTMP) that employ a gene transfer and comply with EU directive definition of organisms are also subject to the Genetically Modified Organisms (GMO)/Genetically Modified Microorganism (GMM) regulatory framework in Europe or Living Modified Organisms in Japan. Currently, two key directives are underlying the GMO regulatory environment for clinical trials in Europe – contained use and deliberate release – of which EU member state-level adaptations introduce even broader regulatory differences.

Figure 2: ATMP clinical trial approval.
Having such a disparate array of requirements leads to differences in submission package content, timelines and other stakeholder responsibilities (eg, site-level certifications and submissions) between the different countries within Europe. In the US and Japan, ATMP-related requirements are also defined,2 with approval from the reviewing authority required prior to the commencement of clinical trials involving the product. In many cases, ATMP authorisation is required before the clinical trial application can be considered. Of note, in general, the above listed challenges tend to accumulate in the startup phase of the clinical trial. Once these challenges are successfully addressed and the clinical study transitions to the conduct phase (once the first patient has been enrolled into the clinical trial), trial execution more closely resembles a non-ATMP clinical trial. More details on the European Regulatory Framework can be found in Dave Li’s 2019 publication.3
Solution: what can be done strategically to improve trial operations?
The startup phase of an ATMP clinical trial constitutes an imperative strategic decision stage of the trial, laying ground for its success. Trial startup activities require not only extensive and comprehensive documentation to be developed for submission purposes and clinical site use (eg, safety instructions), but also demand much broader interactions with competent authorities, study teams, clinical sites and their pharmacy personnel than conventional non-ATMP clinical trials. Due to a strong scientific component underlying these trials, it is necessary to have multidisciplinary experts with relevant scientific backgrounds serving as facilitators and intermediaries in this multi-partite system (agencies-sites-teams) providing subject matter-related support, communication and oversight. One of the crucial and determining due diligences to be performed by these teams is the analysis and identification of the optimal site profile. The analysis must account for the biosafety capabilities of each site, provide proper education to address any perceived trial risks and understand the clinical supply and equipment needs for each site to have success in conducting the trial (Figure 3). If identified as necessary, these experts can also assist in on-site visits to mediate subject matter discussions and training with investigators, site personnel and pharmacies. Another key area where expertise is imperative is the utilisation of regulatory knowledge to leverage the similarities between European countries and local experience to make informed decisions based on study geography and operations.
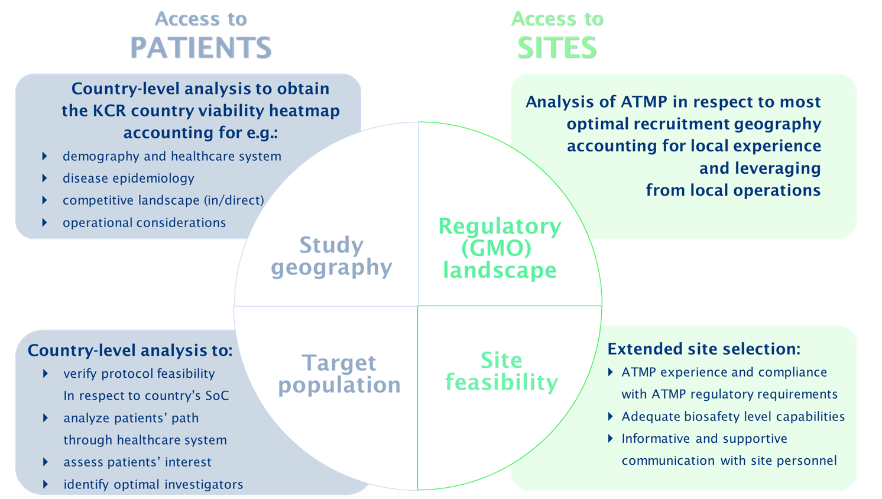
Figure 3: Sample assessment process of access to patients and sites to be performed with engagement of the ATMP expert team.
Conclusion: clinical team expertise is crucial
As briefly outlined above, developers of ATMPs need to confront both challenges related to the clinical trial itself as well as hurdles triggered by the classifications of the investigational medicinal product. Fortunately, with increased exposure to this form of clinical research, competent authorities and clinical sites show increasing familiarity and enthusiasm towards ATMP research. The bridging component in successful and orchestrated trial execution lies with expert guidance and support to help deliver ATMP-related milestones.
About the author
Magdalena Czarnecka has over 15 years of experience in pre-clinical molecular physiology research, health technology assessment and strategic analysis for clinical research development programmes. In her current role at KCR, she is responsible for clinical trial execution strategy and the development and implementation of clinical solutions. Magdalena has also supported ATMP study start-up operations as part of the ATMP Experts Team.
References
- Ten Ham RMT, Hoekman J, Hövels AM, Broekmans AW, Leufkens HGM, Klungel OH. Challenges in Advanced Therapy Medicinal Product Development: A Survey among Companies in Europe. Mol Ther Methods Clin Dev. 2018 Oct 11;11:121-130. doi:10.1016/j.omtm.2018.10.003. eCollection 2018 Dec 14. PubMed PMID: 30456217;PubMed Central PMCID: PMC6234262.
- USA: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research. Determining the need for and content of environmental assessments for gene therapies, vectored vaccines, and related recombinant viral or microbial products guidance for industry. 2015. Japan: JP: Secretariat of the Convention on Biological Diversity. Cartagena Protocol on Biosafety to the Convention on Biological Diversity: Text and Annexes. Montreal: Secretariat of the Convention on Biological Diversity, 2000.
- Li, D. European Regulatory Framework and General Path to Market for ATMPs [Internet]. September 2019 [cited 1 June 2020]. Available from: http://www.appliedclinicaltrialsonline.com/european-regulatory-framework-and-general-path-market-atmps
Related topics
Biologics, Biopharmaceuticals, Clinical Trials, Industry Insight, Regulation & Legislation, Therapeutics
Related organisations
European Union (EU), The European Medicines Agency (EMA), US Food and Drug Administration (FDA)





