

Practical considerations in analysing biologically active peptides by Electrospray Ionisation (ESI) Mass Spectrometry
Posted: 22 October 2013 | | No comments yet
Neuromodulators such as calcitonin gene-related peptide (CGRP) and vasoactive intestinal peptide (VIP) act as biomarkers for pain assessment (pre-clinical). These markers can be detected at low concentrations by Electrospray Ionisation (ESI) Mass Spectrometry (MS). Currently, little is known about the factors affecting responsiveness in the ESI process though the response of peptide ions depends heavily on analyte basicity in solution, pKb, sampling and instrumental conditions. Other factors may be equally important, however, not enough is known about the link between peptide characteristics and selectivity in the ESI process. Here, we review our findings on these links to improve the analyte signal and allow robust quantitation of these important peptide biomarkers…
The measurement of peptides plays a major role in diagnostics for driving the development of new, more specific and sensitive techniques. For example, neuropeptides, a class of chemical messengers that modulate nervous system responses and influence many neurobehavioral functions, could be important markers relevant to the perception and management of pain1.
The detection of peptides can be achieved by MS. The most popular approach to this work is using ionisations techniques such as MALDI or ESI – both soft ionisations methods. For MALDI, the MH+ species is usually the most dominant ion observed in the mass spectrum whereas ESI-MS yields multiple charged ions, [M+nH]n+. In ESI-MS, analytes are introduced into the mass spectrometer in solution. The production of gas phase ions occurs under atmospheric pressure by producing charged droplets at a charged ES capillary tip. The shrinkage of the charged droplets due to solvent evaporation and repeated fission results in highly charged gas phase ions (protonation of the basic amino acids (Lys, Arg, His) and of the N-terminus) which can be separated according to their mass-to-charge ratio (m/z). To achieve protonation, the analytes must be basic both in solution (low pH) and in the gas phase. It is often expected that the primary sequence of the analytes and the pKb of the constituent ionisable amino groups of the peptide will be prominent factors in determining the number of protonation during positive ESI-MS analysis 2,3. However, this is not always the case4.
Although ESI mechanisms are still debated it is well-established that the observed ESI response (selectivity, charge state distribution (CSD) and signal intensity of ions in the resulting mass spectrum), is affected by instrumental parameters such as sheath gas flow rate5 or ES source geometry6, by analyte concentration, solvent and analyte basicity7-9, solvent surface tension10,11, gas phase basicity12 and pH13. Further, properties such as number and type of amino acids and protein conformation7,13,14 also play a role in determining the maximum obtainable charge state and CSD. For peptides, which have little secondary and tertiary structure, CSDs are independent of peptide secondary structure15,16.
Practical considerations – developing a deeper understanding
The focus of our research is the development of a deeper understanding of the factors that affect selectivity, CSD and ion suppression during positive ESI-MS analysis of peptides. In this short review, we will share some interesting observations.
Additives are usually added to the mobile phase to influence analyte ionisation, improve the ion signal and achieve conditions convenient for the sake of interfacing ESI-MS and chromatographic separation (LC-MS). When analysing peptides (ESI positive mode), the mobile phase is usually acidified (typically 0.1%v/v) with formic acid (FA), acetic acid or ammonium hydroxide. Increasing the acid concentration will, as expected, increase the charge state on the dominant ion in the mass spectrum and potentially push the CSD towards higher values (lower m/z) as the solution pH drops below the pKa of the acidic side chains in the peptide. In addition, increasing the acid concentration will initially generate a greater number of protonated ions and potentially result in lower m/z ions because of an increasing number of available protons. However, as we observed, above a critical analyte to acid ratio, ion suppression occurred resulting in poor signal intensities8,17. This means that more intense ion signals are observed at higher pH values than at lower pH values where the higher charge state species are observed. For example, for VIP (3,326 Da), the intensities of the [M+4H]4+ and [M+5H]5+ species were greater at mobile phase pH 3.23 than the intensity of the [M+6H]6+ species at pH 2.3.
Generally, decreasing the mobile phase flow rate also increased the intensity of the observed base peak ion. The best ESI sensitivities are usually obtained in the absence of a nebuliser gas18. When conventional μL/min flow rates are employed, the ESI response is characterised as a ‘concentration‐sensitive’ technique; i.e. the ESI response (intensity) is proportional to the concentration of the analyte and, therefore, slight changes in the flow rate will have no significant effect on the signal intensity19. At extremely low flow rates (~10–100 nL/min), splitting of droplets is presumably readily achieved and the ionisation efficiency can approach 100 per cent; here the ESI response is described as proportional to the absolute quantity20.
As expected, increasing the analyte concentration resulted in increased competition for available protons, and hence a decrease in the appearance of lower m/z ions21 i.e. [M+5H]5+< [M+4H]4+ < [M+3H]3+ < [M+2H]2+. We found a linear correlation between analyte concentration and ESI response for the protonated states with higher charge states dominant at low analyte concentrations (1–500 ng/mL, flow rate 300 nL/min) and lower charge states dominant at higher concentrations (500–5000 ng/mL), suggesting insufficient availability of protons as the analyte concentrations increased8.
The effect of mobile phase composition was found to be very powerful. In addition to solubilising the peptides prior to ionisation, the most important role of the solvent is charge stabilisation, and solvent evaporation from charged ESI droplets causes shrinkage and splitting of droplets. The latter is reviewed by Kebarle & Verkerk and the dependence on the type of solvent is considered relatively small22. Though the correlation between the solution and gas phase properties is still being debated, several studies have shown that solution characteristics are retained in vacuo23-25. This means that initial solvent conditions define the starting conditions and the conformation of the peptides. Studies by Konermann & Douglas and Babu & Douglas have shown that the CSDs are unaffected by changes in the proteins secondary structure but only to changes in the tertiary structure15,16. In line with this, and based on Hydrogen / Deuterium exchange experiments, we concluded that all protonated sites are readily exchanged and so any conformation of these peptides does not affect observed CSDs.
Interestingly, the analysis of one peptide (Prolactin-releasing peptide, PrRP, 2,273 Da) revealed that the observed dominant charge states, [M+3H]3+ and [M+4H]4+ were affected by the relative volatility of the mobile phase: in mixtures (with H2O and FA) containing high boiling point solvents (IPA, 88.15°C or MeCN 81.6°C), the [M+3H]3+ ion dominated, however in mixtures with lower boiling point solvents (DCM 39.6°C or MeOH 65°C), the [M+4H]4+ ion dominated. This means that for PrRP, a higher charge state dominates in more volatile solvent mixtures. This could be attributed to a more rapid desolvation of the droplets: MeOH or DCM (in H2O and FA) evaporate faster than droplets with IPA or MeCN because of their higher vapour pressure. Interestingly, studies by Sterner and co-workers conclude that the maximum charge state might be more dependent on the proton affinity of the less volatile solvent26; there is a large loss of solvents before the small droplets containing the peptides are formed. However, for most of the analysed peptides the solvent environment did not result in a change in the observed dominant charge state. Hence, solvent volatility, by itself, is not sufficient to affect overall charge state. However, the intensities of the ions in the ESI+ mass spectra are affected by the solvent used: in protic solvents (MeOH or isopropanol) mixtures (25, 50 or 75%v/v, with H2O and FA) the ion intensities were significantly higher than in spectra collected in aprotic solvents (MeCN or DCM) mixtures. Therefore, the ESI responses indicate that acetonitrile might not be the best solvent for ESI-MS analysis of neuropeptides and although they are detected as positive ions, these peptides are better analysed in MeOH or IPA mixtures.
Effects induced by the electrospray conditions cannot be ruled out and the use of a nano ESI source would minimise / remove this effect. In line with our observations, Iavarone and co-workers reported that small peptide ions were observed to deprotonate when small amounts of solvent less volatile than water were added to the droplets27. However, they show the opposite for proteins: it is possible to enhance ESI charging of proteins (‘supercharge’) by applying high surface tension or addition of solvents with relatively low volatility. They rationalised the observation in terms of solvent composition and number of gas phase collisions affecting the proton transfer reaction in the ionisation process. When the supercharging reagent m-NBA (0.1 % v/v) was added into mobile phase solvents (0.1%FA and MeCN+0.1% FA with the acetonitrile volume at 25, 50 and 75% v/v), two trends were clear. Firstly, an increase in the acetonitrile concentration in the presence of m-NBA caused an increase in the ESI response (intensity). This was a very interesting observation as for these peptides – in the absence of m-NBA – ion suppression is observed when the mobile phase becomes increasingly more non-polar (increasing the concentration of MeCN in mobile phases from 10 – 90 per cent), for example in Figure 1. In water:MeCN mixtures, the analytes, depending on their polarity (estimated based on28), will remain in the decreasing aqueous interior of the droplets where they are less able to compete for the excess charge at the droplet/air interphase in the ESI process. The less polar peptides are expected to ionise better compared to the more net polar peptides as they will be able to remain on the droplet / air interphase for longer, taking up the excess charge. The more non-polar peptides, which carry a greater fraction of the excess charge, will give a higher ESI response17,19,21,29. This is also clear from the data. In the presence of 0.1 per cent m-NBA and with increasing concentration of organic content in the mobile phase, the greatest improvement in base peak ion intensity is indeed observed for the most non-polar peptides. Secondly, the data show that higher charge state ions are formed when relatively small amounts (0.1 per cent) of m-NBA were added to the typical mobile phase solutions of varying acetonitrile concentrations. The CSD increased by one unit for all peptides analysed. The increase in percentage m-NBA in the mobile phase to 0.7 per cent did not push the charge state distribution towards higher values. The reason for this is that the peptides only contain a certain number of basic amino residues (including the N-terminal) which can be protonated and we concluded that the C-terminal for these peptides, in spite of the solution pH, was protonated (COOH).
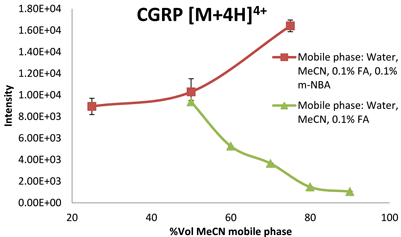
Figure 1: ESI response of the quadruply charged species of CGRP (3,126 Da) in the presence and absence of m-NBA in mobile phases with increasing acetonitrile concentrations %v/v
Another interesting observation relating to the amino acid composition and peptide polarity was that responsiveness is determined according to the presence of terminal arginine > most non-polar > most polar peptide. Arginine at either the C- or N-terminus enhances the electronegativity of the peptide and is a source of selectivity during positive ESI analysis. Our data from MALDI analysis supports that of the ESI experiments; peptides with a terminal arginine residue led to higher signal intensities. However, in peptide mixtures, peptides with a C-terminal arginine always dominated in the mass spectrum7.
We have also studied the effect of adding electrolytes to the mobile phase. It is possible to decrease the charge state of the dominant ion by addition of ammonium acetate. In the presence of increasing amounts of ammonium acetate (10-7 to 10-2 M), the observed CSD of most investigated peptides were pushed towards lower levels in line with observations made by Wang & Cole30 and Mirza & Chait31. The change in ion ratios and, for some analytes, the reduction in total ion intensity indicates that the presence of acetate counter ions favours the removal of protons from the peptides or neutralisation of positive charge sites in the peptide. Other non-volatile salts (sodium chloride and sodium bicarbonate) resulted in electrolyte-analyte cluster formation only.
Conclusions
The ESI response can be affected by many factors and in this short review, the effect of choice of solvent, pH, analyte concentration, flow rate, solvent composition (including additives such as m-NBA and electrolytes), analyte polarity and position of amino acids in the terminal regions was summarised. The findings extend our understanding of the ESI+ process and provide a rational approach to optimising sensitivity of the electrospray conditions where a narrow mass range of peptides is poorly chromatographically resolved. The information contributes to more effective method development, especially during label-free quantitative determination of peptides extracted in solution.
Acknowledgements
The author would like to thank past and present members of the mass spectrometry group at Greenwich University.
Biography

Birthe V. Nielsen, Faculty of Engineering and Science, University of Greenwich
Dr Birthe Nielsen obtained her MSc in Biotechnology from Aalborg University and continued with an industry-funded PhD in the UK. In 2007, Dr Nielsen joined the University of Greenwich, where she has been pursuing her own research interests in mass spectrometry, building a deeper understanding of how certain peptides ionise and behave during Electrospray Ionisation.
Email: [email protected]
References
- Fischer HP, E. W., Russell IJ., A possible role for saliva as a diagnostic fluid in patients with chronic pain. Semin Arthritis Rheum. 1998, 27, 348-59
- Ikonomou, M. G.; Blades, A. T.; Kebarle, P., investigations of the electrospray interface for liquid-chromatography mass-spectrometry. Analytical Chemistry 1990, 62, 957-967
- Ehrmann, B. M.; Henriksen, T.; Cech, N. B., Relative importance of basicity in the gas phase and in solution for determining selectivity in electrospray ionization mass spectrometry. Journal of the American Society for Mass Spectrometry 2008, 19, 719-728
- Mansoori, B. A.; Volmer, D. A.; Boyd, R. K., ‘Wrong-way-round’ electrospray ionization of amino acids. Rapid Communications in Mass Spectrometry 1997, 11, 1120-1130
- Girod, M.; Dagany, X.; Antoine, R.; Dugourd, P., Relation between charge state distributions of peptide anions and pH changes in the electrospray plume. A mass spectrometry and optical spectroscopy investigation. International Journal of Mass Spectrometry 2011, 308, 41-48
- Winger, B. E.; Lightwahl, K. J.; Smith, R. D., Gas-phase proton-transfer reactions involving multiply charged cytochrome-c ions and water under thermal conditions. Journal of the American Society for Mass Spectrometry 1992, 3, 624-630
- Abaye, D. A.; Pullen, F. S.; Nielsen, B. V., Peptide polarity and the position of arginine as sources of selectivity during positive electrospray ionisation mass spectrometry. Rapid Communications in Mass Spectrometry 2011a, 25, 3597-3608
- Abaye, D. A.; Pullen, F. S.; Nielsen, B. V., Practical considerations in analysing neuropeptides, calcitonin gene-related peptide and vasoactive intestinal peptide, by nano-electrospray ionisation and quadrupole time-of-flight mass spectrometry: monitoring multiple protonations. Rapid Communications in Mass Spectrometry 2011b, 25, 1107-1116
- Tang, L.; Kebarle, P., Dependence of ion intensity in electrospray mass-spectrometry on the concentration of the analytes in the electrosprayed solution. Analytical Chemistry 1993, 65, 3654-3668
- Iavarone, A. T.; Jurchen, J. C.; Williams, E. R., Supercharged protein and peptide lone formed by electrospray ionization. Analytical Chemistry 2001, 73, 1455-1460
- Iavarone, A. T.; Williams, E. R., Mechanism of charging and supercharging molecules in electrospray ionization. Journal of the American Chemical Society 2003, 125, 2319-2327
- Carr, S. R.; Cassady, C. J., Reactivity and gas-phase acidity determinations of small peptide ions consisting of 11 to 14 amino acid residues. Journal of Mass Spectrometry 1997, 32, 959-967
- Chowdhury, S. K.; Katta, V.; Chait, B. T., Probing conformational changes in proteins by mass spectrometry. Journal of the American Chemical Society 1990, 112, 9012-9013
- Kaltashov, I. A.; Abzalimov, R. R., Do ionic charges in ESI MS provide useful information on macromolecular structure? Journal of the American Society for Mass Spectrometry 2008, 19, 1239-1246
- Konermann, L.; Douglas, D. J., Unfolding of proteins monitored by electrospray ionization mass spectrometry: A comparison of positive and negative ion modes. Journal of the American Society for Mass Spectrometry 1998, 9
- Babu, K. R.; Moradian, A.; Douglas, D. J., The methanol-induced conformational transitions of beta-lactoglobulin, cytochrome c, and ubiquitin at low pH: A study by electrospray ionization mass spectrometry. Journal of the American Society for Mass Spectrometry 2001, 12
- Kelly, M. A.; Vestling, M. M.; Fenselau, C. C.; Smith, P. B., Electrospray analysis of proteins – a comparison of positive-ion and negative-ion mass-spectra at high and low pH. Organic Mass Spectrometry 1992, 27, 1143-1147
- Karas, M.; Bahr, U.; Dulcks, T., Nano-electrospray ionization mass spectrometry: addressing analytical problems beyond routine. Fresenius Journal of Analytical Chemistry 2000, 366, 669-676
- Cole, R. B., Some tenets pertaining to electrospray ionization mass spectrometry. Journal of Mass Spectrometry 2000, 35, 763-772
- Covey, T., Biochemical and Biotechnological Applications of Electrospray Ionization Mass Spectrometry. In Snyder, A. P., Ed. American Chemical Society: Washington, DC, 1995; pp 21-59
- Cech, N. B.; Enke, C. G., Relating electrospray ionization response to nonpolar character of small peptides. Analytical Chemistry 2000, 72, 2717-2723
- Kebarle, P.; Verkerk, U. H., Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrometry Reviews 2009, 28, 898-917
- Chung, E. W.; Nettleton, E. J.; Morgan, C. J.; Gross, M.; Miranker, A.; Radford, S. E.; Dobson, C. M.; Robinson, C. V., Hydrogen exchange properties of proteins in native and denatured states monitored by mass spectrometry and NMR. Protein Science 1997, 6, 1316-1324
- Jorgensen, T. J. D.; Roepstorff, P.; Heck, A. J. R., Direct determination of solution binding constants for noncovalent complexes between bacterial cell wall peptide analogues and vancomycin group antibiotics by electrospray ionization mass spectrometry. Analytical Chemistry 1998, 70, 4427-4432
- Lim, H. K.; Hsieh, Y. L.; Ganem, B.; Henion, J., Recognition of cell-wall peptide ligands by vancomycin group antibiotics – studies using ion-spray mass-spectrometry. Journal of Mass Spectrometry 1995, 30, 708-714
- Sterner, J. L.; Johnston, M. V.; Nicol, G. R.; Ridge, D. P., Apparent proton affinities of highly charged peptide ions. Journal of the American Society for Mass Spectrometry 1999, 10, 483-491
- Iavarone, A. T.; Jurchen, J. C.; Williams, E. R., Effects of solvent on the maximum charge state and charge state distribution of protein ions produced by electrospray ionization. Journal of the American Society for Mass Spectrometry 2000, 11, 976-985
- Kyte, J.; Doolittle, R. F., A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology 1982, 157, 105-132
- Wang, G.; Cole, R. B., Wang, G. Cole, R.B. In Electrospray Ionization mass Spectrometry: Fundamentals, Instrumentation and Applications, Cole, R. B., Ed. John Wiley: New York, 1997; pp 137-174
- Wang, G. D.; Cole, R. B., Effect of solution ionic-strength on analyte charge-state distributions in positive and negative-ion electrospray mass-spectrometry. Analytical Chemistry 1994, 66, 3702-3708
- Mirza, U. A.; Chait, B. T., Effects of anions on the positive-ion electrospray-ionization mass-spectra of peptides and proteins. Analytical Chemistry 1994, 66, 2898-2904
Issue
Related topics
Electrospray Ionisation (ESI) Mass Spectrometry, Mass Spectrometry

