Application of LCMS in small-molecule drug development
Posted: 24 August 2016 | Chunang (Christine) Gu (Genentech), David Russell (Genentech), Peter Yehl (Genentech) | No comments yet
As a technology, mass spectrometry (MS) has evolved to the point where it is used throughout the drug development process. In particular, when MS is coupled with high-performance liquid chromatography (HPLC) it adds an orthogonal detection function for sample analysis and provides unique capabilities for pharmaceutical analysis, such as sensitivity, selectivity, speed of analysis and rich mass information. This article describes the fundamentals and general applications of hyphenated LC-MS in supporting small-molecule drug development, such as impurity and degradant structural characterisation, and qualitative and quantitative analysis. In addition, some newly-developed technologies in LC and MS are discussed in terms of their future application within pharmaceutical development.

Introduction to LC-MS
LC is a separation technique that relies on analytes’ differences in partitioning behaviour between a flowing mobile phase and a stationary phase to separate the components in a mixture. For most small-molecule drug development, LC is now one of the most powerful tools in analytical chemistry1,2. MS uses the difference in mass-to-charge ratio (m/z) of ionised compounds to separate them from each other. It is often considered as the most sensitive detector and is typically coupled with other technologies, most commonly LC. This type of orthogonal-mass spectrometric methodology has facilitated drug development enormously, primarily due to the superior speed, sensitivity and selectivity of such ‘hyphenated’ techniques. At present there are four major stages of drug development: drug discovery, preclinical development, clinical development and manufacturing3 . The small-molecule drug’s early development is also known as chemistry, manufacturing and control (CMC).
Ionisation and LC-MS general applications
The primary difficulties of combining LC and MS have been the interface, the ionisation of analytes in a stream of condensed liquids and transfer of the ions into the high vacuum inside the mass spectrometer. The most common forms of ionisation modes coupled with LC in small-molecule research are electrospray ionisation (ESI); atmospheric pressure chemical ionisation (APCI); and atmospheric pressure photoionisation (APPI). As illustrated in Figure 1 ESI is better suited to higher-molecular-weight and polar compounds, while APCI is best suited for low- to medium-polarity compounds with a limited upper mass range (< m/z of 1,000).
Recent studies have shown that APPI expands the range of compounds that can be ionised and has become the third option of choice for less polar compounds, such as steroids and PAHs4,5.
Electron ionisation (EI) is typically used in gas chromatography-MS for small and volatile molecules. Aviv A. et al. have described an approach to bring EI back to LC-MS based on sample ionisation as vibrational cold molecules in a supersonic molecular beam (Cold EI)6 . Cold EI results in enhanced molecular ions that are often weak or missing in standard EI, however the technique is fully compatible with library identification. This renovated EI-LC-MS technique has great potential to identify unknown samples that cannot be ionised by ‘soft ionisation’ sources.
LC-MS for drug impurity identification and profiling
Impurity identification and profiling is critical to the assurance of patient safety and drug efficacy in a drug development and active pharmaceutical ingredient (API) manufacturing unit. Regulatory authorities have established clear and rigorous guidelines which dictate the identification of impurities at lower levels, depending upon dosage7,8. For most small-molecule drug-like compounds, LC is by far the most reliable and efficient separation technique, and serves as the main workhorse for impurity profiling.
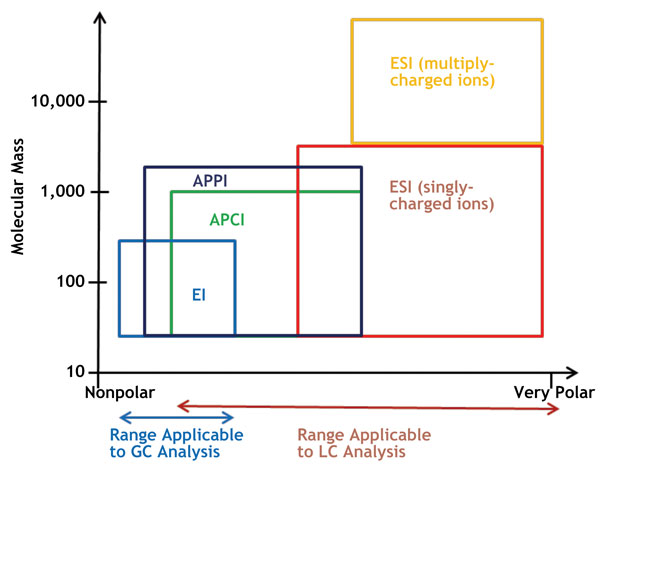
Figure 1: Common ionisation techniques and application areas
The orthogonal method of coupling MS with LC provides unique solutions to a wide range of structural characterisation problems for impurity profiling. It not only addresses relevant regulatory issues, but also provides clues to further process optimisation during both drug substance and drug product development. A general MS-based strategy to analyse small-molecule impurity and degradants is shown in Figure 2.
At the early stages of drug development, rapid analysis methods that provide nominal molecular weight data along with isotopic distribution pattern are commonly used. As a project progresses through clinical development, the structures of unknown impurities are required and nominal mass measurements are no longer sufficient to elucidate these structures with confidence. Accurate mass is used to determine elemental compositions of impurity structures, an essential step in elucidating the structures of unknown compounds. There are several different types of MS analysers capable of providing accurate masses, including magnetic sector, time-of-flight (TOF), orbital trap and Fourier transform-ion cyclotron resonance (FT-ICR) systems. In addition to advanced instrumentation, software can also help extend nominal mass data to high-resolution data by using a post-acquisition approach to calibrate mass spectral accuracy9.
Additional structural information can be obtained from tandem-intime/space MS instruments, such as ion trap, triple-quadrupole and Qtrap systems. The molecular ions are fragmented within the mass analyser and the resulting neutral losses by multiple-stage MS fragmentation (MSn) processes are informative for structure elucidation of various chemical/functional groups on target molecules. This greatly facilitates the understanding of the ion fragmentation pathway for an unknown species and enables the identification of unknown compounds. Moreover, accurate mass data on fragment ions, which can be generated by high-resolution MS/MS systems, provide additional evidence to support structural assignments.
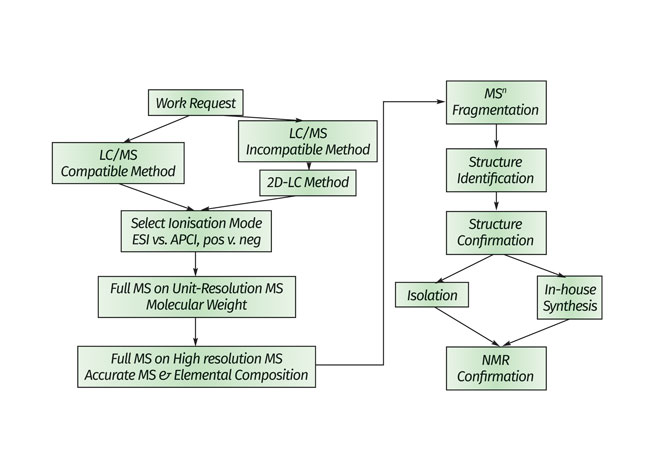
Figure 2: MS-based strategy for impurities’ and degradants’ identification
One challenge in elucidating the structure of unknown compounds using LC-MS is that non-volatile buffers, which are not amenable to MS ionisation, are often required for isolation of the compound of interest. As shown in the MS-based workflow (Figure 2), two dimensional-LC-MS can often be used to overcome this issue and has the added advantage of improved chromato graphic resolution10,11. The first LC dimension utilises the non MS- compatible LC isolation method to achieve the desired chromato graphic performance. The analytes of interest are stored in loops/vials. The second dimension then uses MS-compatible solvents to deliver the isolated analytes from the first dimension to the mass spectrometer for structural analysis.
Quantitative analysis of trace level small molecules
Coupled with LC, MS has become the detector of choice for superior sensitivity and selectivity in pharmaceutical compound quantification analysis. The combination of superior performance and ease of use has led to widespread adoption of single-quadrupole LC-MS systems in regulated laboratories. However, with the recent development in novel therapies and more stringent regulatory requirements, single quadrupole LC-MS cannot always meet the needs of small-molecule drug development. The use of a triple-quadrupole LC-MS/MS (LC-QqQ-MS) has become prevalent in small-molecule quantitation analysis due to its high sensitivity.
The most common method used in MS/MS quantitation is multiple reactions monitoring (MRM), which selects a parent ion in Q1 and monitors its unique fragment ion in Q3. The latest LC-QqQ-MS system can detect impurities well below the limits required by regulatory authorities in potential genotoxic impurities (PGIs). This is illustrated in Figure 3A which shows how simultaneous analysis of four PGIs for one API was achieved by using LC-MS/MS in MRM mode. The detection limits for these four PGIs on single-quadrupole LC-MS were about 1μg/mL (data not shown) due to the presence of high interference noise in the spectral background. By moving to LC-QqQ-MS, the sensitivity of the impurities were increased approximately 1,000 times – a sufficient amount to perform the limit test for trace level impurities during the purging study in early drug development.
Cleaning verification tests also demand highly sensitive analytical methods. These tests are typically performed to ascertain the effectiveness of the procedure used to clean pharmaceutical processing equipment such as blenders and tablet presses. Sensitivity and free-from-interfering peaks are the most critical requirements. The LC-MS/MS method is well established as a versatile tool for quantifying known compounds in the solvent rinsates, or swabbing extracts from manufacturing equipment12. This is especially useful when dealing with cleanout testing for high potency drugs, i.e., human health criteria category three and four compounds, where the acceptance criteria requires low ng/mL detection.
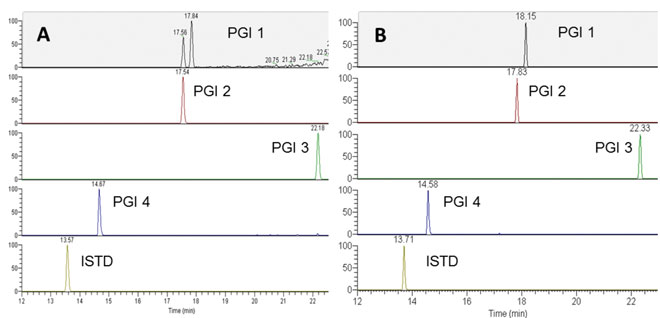
Figure 3: Chromatograms of 4ng/mL of PGIs spiked into 4mg/mL of API. (A) The data were
acquired on LC-QqQ-MS instrument. (B) The data were acquired on a high-resolution MS
instrument. Reproduced with permission from Gu et al., American Pharmaceutical Review (2015)
Recently, LC-high resolution MS (HRMS) has shown promise as a sensitive, accurate, reproducible and reliable alternative to the use of LC-QqQ-MS in a quantitation study13-15. In full-scan HRMS experiments for small-molecule quantification, selectivity is achieved by creation of extracted ion chromatograms of quasimolecular ions of the compound of interest, with a narrow mass-extraction window. The more narrow the setting of the mass-extraction window, the higher the selectivity. This is illustrated in Figure 3B: the chromatogram of four PGIs was acquired on a HRMS instrument at full scan mode and the data were processed by extraction of the signal from compounds with a protonated mass-tocharge ratio within a five part-per-million mass accuracy window. Compared with traditional LC-QqQ-MS, sensitivity and selectivity do not significantly drop with the HRMS system, and the response is linear which enables reliable quantitation.
An additional attractive benefit of using HRMS is that selection of a specific precursor ion is not required in the full scan experiment. Therefore, the experiment simplifies the MS method set-up and saves scientists’ development time. Moreover, researchers have the flexibility to mine full data sets at a later date and there is far greater information from each sample16
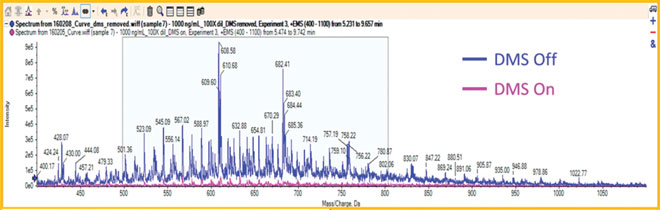
Figure 4: Mass spectra of the elution from a shielded hydrophobic phase column with DMS and without DMS
Future perspectives and conclusions
The wide applicability of LC-MS in the pharmaceutical industry has been achieved by the continuous improvements in LC and MS, respectively. First and foremost was the advent and trans formation of HPLC to ultrahigh pressure liquid chromatography (UHPLC). High efficiency UHPLC separation with increased theoretical plates can allow significant improvements in peak capacity and has the ability to shorten run times considerably. The consequences for MS coupling with UHPLC, however, are that very narrow chromatographic zones require high scanning speed to avoid the loss of chromato graphic resolution. It is still a challenge for high-resolution, accurate mass systems to provide a sufficient number of scans (≥10) across the chromatographic peak in full scan mode without compromising resolving power (resolution) and sensitivity.
The time-of-flight MS (TOF) platform is well known for its rapid scanning rate. Newly-developed high-field orbitrap mass analyser geometry also improves scan speeds four fold17, making the orbitrap more compatible with UHPLC separations. The latest developments in MS also help take full advantage of LC, making this technology more accessible to pharmaceutical research. For example, a shielded hydrophobic phase (SHP) column allows untreated biological samples to be directly injected onto the column and avoids the time-consuming sample cleanup step18,19. The SHP column consists of a polymeric surface containing hydrophobic pockets enclaved by a hydrophilic network. Small molecules penetrate the water-solvated interface of the hydrophilic network to interact with the hydrophobic regions. The hydrophilic shielding prevents larger water-solvated molecules, such as protein, from entering into interactions with hydrophobic groups.
Recently SHP columns have been widely used to analyse the unconjugated drug in the presence of proteins and protein-drug conjugates20,21. However, the SHP column is not suitable for MS detection because polyethylene oxide bleeds from SHP packing material. Polyethylene oxide is readily ionised and causes high background noise in the spectra when coupled with LC-MS (blue trace in Figure 4). With the newly developed ion mobility technique coupled with the mass spectrometer (IMS)22, it can help remove the column bleed species from the compounds of interest and serve as a mass filter. When the different compensation voltage was applied on the ion mobility system, the background from polyethylene oxide was significantly removed (purple trace in Figure 4). The method of coupling the SHP column with differential ion mobility MS (DMS) demonstrated the good specificity and sensitivity in detecting the trace level of free drug species in the conjugate samples without sample preparation.
This article highlights the orthogonal method of combining the high resolving power of LC with the superior mass detection capacity of MS for small molecule characterisation and quantitation. With continual improvement in ionisation methods, column chemistry, software and instrumentation, LC-MS will gain a widened scope of application and increasingly penetrate the various stages of drug discovery and development.
About the author
Christine Gu is currently a Scientist in the Department of Small Molecule Pharmaceutical Science at Genentech. She is a subject matter expert in mass spectrometry. Before joining Genentech, she worked as a Senior Application Scientist at ThermoFisher Scientific. She holds a PhD degree in Toxicology at University of California, Riverside.
Issue
Related topics
Drug Development, HPLC, Liquid Chromatography - Mass Spectrometry (LC-MS), Mass Spectrometry