

Establishing assays and small molecule screening facilities for Drug discovery programs
Posted: 16 February 2011 |
Although many of the marketed small molecule drugs have been discovered by research and development efforts within the pharmaceutical industry, there has been a paradigm shift with external sources increasingly being relied upon to fill their pipelines. This trend is likely to increase and the key pre-clinical activities carried out by organisations outside the pharmaceutical industry include target validation, assay development and their use in High Throughput Screening campaigns, validation of the Hit molecules, Hit-to-Lead and Lead-to-Candidate screening/chemistry. In order to perform these activities, adequate know-how and technical expertise is essential so that the processes meet appropriate industry standards. This article discusses some of the challenges associated with assay development and the automation of High Throughput Screening.
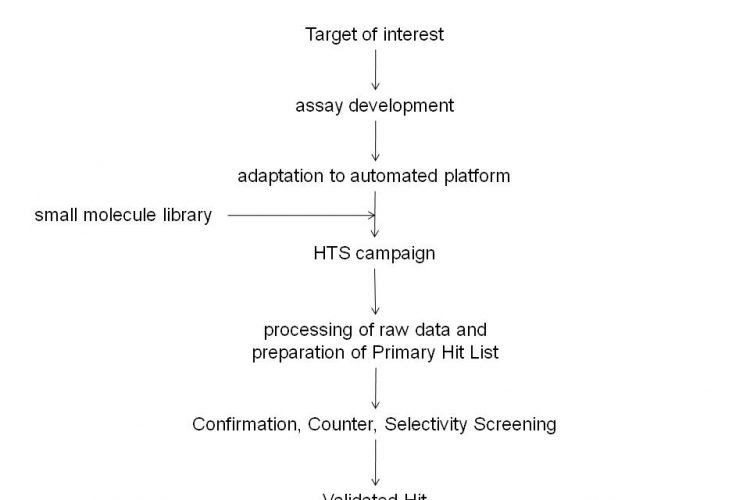
Figure 1 Typical critical path for small molecule Drug Discovery programs. Drug Discovery involves identification of a Target of interest for which an assay is developed. This assay is then adapted for screening purposes and utilised in a High Throughput Screening campaign against small molecule libraries. The High Throughput Screening campaign will usually yield many Hit molecules. Confirmation, Counter and Selectivity Screening will provide a final list of Validated Hits.
Although many of the marketed small molecule drugs have been discovered by research and development efforts within the pharmaceutical industry, there has been a paradigm shift with external sources increasingly being relied upon to fill their pipelines. This trend is likely to increase and the key pre-clinical activities carried out by organisations outside the pharmaceutical industry include target validation, assay development and their use in High Throughput Screening campaigns, validation of the Hit molecules, Hit-to-Lead and Lead-to-Candidate screening/chemistry. In order to perform these activities, adequate know-how and technical expertise is essential so that the processes meet appropriate industry standards. This article discusses some of the challenges associated with assay development and the automation of High Throughput Screening.
Since the completion of the sequencing of the human genome and the subsequent identification of the many thousands of genes encoded by it, the pharmaceutical industry has expended a significant effort in attempting to identify the products of genes which are associated with disease processes1-6. These targets would then be explored for drug discovery purposes and a series of activities would be initiated which would include the preparation of biological reagents that contain these targets and the development of assays which would subsequently be utilised in High Throughput Screening (HTS) campaigns against libraries of small molecules to identify chemical starting points for drug discovery7,8. When initiating a drug discovery program that will make use of an HTS campaign, it is essential to have in place a critical path to allow its appropriate progression (see Figure 1). The primary hits that are identified from the HTS campaign should be evaluated in suitable counter assays (to identify artefacts due to interference in HTS assays), selectivity assays (additional targets against which the activity of compounds should be determined) and secondary assays (typically cell based assays that make use of the target in its native environment). In addition, the liability profile of compounds in P450, HERG, PGP assays, the in vivo profile and physico-chemical properties would be determined in order to allow their prioritisation for further study.

Figure 1 Typical critical path for small molecule Drug Discovery programs. Drug Discovery involves identification of a Target of interest for which an assay is developed. This assay is then adapted for screening purposes and utilised in a High Throughput Screening campaign against small molecule libraries. The High Throughput Screening campaign will usually yield many Hit molecules. Confirmation, Counter and Selectivity Screening will provide a final list of Validated Hits.
It is now well established that, despite the substantial increase in the number of targets being investigated for drug discovery purposes, there has not been an associated increase in the number of drugs being approved by the regulatory authorities and thus new strategies are being explored to overcome this lack of productivity9,10. A major issue has been that although drug discovery programs deliver potent small molecules that act upon the target of interest in a desired manner, they often fail to exhibit efficacy in clinical trials, thus suggesting that target validation was insufficient. Therefore, the discovery of chemical probes to validate targets should be a pre-requisite to pursuing them for drug discovery purposes. These chemical probes would have defined properties, such as exhibiting appropriate potency for modulating the activity of the target of interest, suitable selectivity and be cell-permeable for use in basic and applied research11. For novel targets in particular, such chemical probes are likely not to exist and their use to validate targets would be expected to de-risk them for drug discovery purposes. Therefore, it may be necessary to re-position HTS and be more realistic with what its output can be used for with less emphasis placed upon it to deliver chemical starting points in drug discovery12.
Assay development
A typical drug discovery program makes use of a molecular target based assay that is compatible with microtitre plates13. The type of assay that is chosen depends on the Target of interest and vendors can be very helpful in providing trial packs of reagents as well as technical support to facilitate rapid assay development. For popular targets, it may be possible to purchase off-theshelf assay kits and this is well exemplified for the kinase target class e.g. Molecular Devices have developed the immobilised metal affinity for phosphochemicals (IMAP®) functional assay for a variety of serine / threonine and receptor tyrosine kinases14. An alternative binding assay for kinases (LanthaScreen™)15-17 is also available from Life Technologies. As a wide choice of assays are available for most target classes, it would be appropriate to develop screening compatible assays in parallel for the target of interest in different formats, use all of them to evaluate any available standard compounds and select the most appropriate assay for progression to the HTS campaign itself. These additional assays that would have been developed would still be of use as counter assays to identify format specific compounds which are potentially artefacts. The availability of both functional and binding assays also offers the ability to provide information about compound mechanism of action. The typical timescales associated with assay development are two to six months however; this could be longer if scale-up of a particular reagent is required. Having successfully developed an assay for the target of interest, it is usually miniaturised, its sensitivity towards DMSO is determined and then it is validated to ensure it meets the criteria for use in the HTS campaign18. It is also necessary to calculate the quantities of any specific reagents that will be required not only for the entire HTS campaign, but also for any necessary experimental follow-up work, which would include confirmation screening, counter screening, mechanism of action studies and any other work that is deemed necessary. It is therefore advisable to prepare a single batch of reagent that will be able to sustain the entire duration of the drug discovery program.
Small molecule libraries
The small molecule libraries that assays can be screened against vary considerably in size from a few thousand (in the case of focused small molecule libraries) through to millions (in the case of diverse small molecule libraries)19-24. Most HTS campaigns are performed using suitably miniaturised assays in 384 or 1,536 well microtitre plate format. The volume of a typical 384 well assay ranges between 10 – 50 μl and in the case of a 1,536 well assay between 4 – 8 μl. As small molecule libraries are commonly stored in 100 per cent v/v DMSO solutions at a concentration of 10 millimetres, in order to ensure that the DMSO concentration in HTS assays is kept suitably low (typically < one per cent v/v), small volumes of the stock compound solutions need to be added per well, therefore appropriate equipment is necessary for this.
Access to small molecule libraries is possible by a variety of methods such as their outright purchase, access on a fee for service basis from a supplier or as collaboration with an organisation that houses such libraries.
High Throughput Screening
Unless the numbers of compounds that are to be screened are low, there are a number of reasons why it is not feasible to use 96 well microtitre plates for HTS campaigns. These include the fact that such plates would typically only allow the evaluation of 80 compounds per microtitre and that reagent consumption and the time taken for the completion of the screening campaign would be prohibitive. Performing HTS campaigns in either 384 or 1,536 well plates is more efficient; however, the final decision on plate density needs to be made on a case-by-case basis. However, it is noteworthy that as assays are miniaturised, reagent consumption and the amount of compound decreases thus allowing for significant cost savings25-27.
A common sense approach should be taken when executing an HTS campaign, in that when screening focused sets of compounds (typically <10,000 compounds) it may be entirely reasonable to employ a manual or semi-automated methodology. For most HTS campaigns, compounds are added first to microtitre plates and significant advances have been made with the hardware used for this, in particular the Labcyte ECHO that makes use of Acoustic Droplet Ejection28,29. This technology has a proven track record in relation to (1) versatile compound dispense volume, (2) dispensation of compound into both 384 and 1,536 microplates, (3) cost savings as no disposable tips are required, (4) no DMSO waste generation, (5) no cross contamination of compounds and (6) on-line QC to verify that compound has been successfully dispensed. Having dispensed compound, the addition of other reagents can take place and this usually has two or three steps. The first step will be the addition of the target of interest to the microtitre plates e.g. protein or cells. In the case of adding protein to the microtitre plates, after a defined pre-incubation step where the compound has the opportunity to have an effect upon the target, addition of substrate / ligand takes place. The final detection step will allow the quantification of the extent to which the compound has modulated target activity and this is performed using appropriate data analysis software30,31. Although the effect of the compound can be represented as a percentage effect, its mechanism of action at this stage would be unknown.
The assay development and HTS infrastructure at the European ScreeningPort is versatile (see Figure 2) and it also contains a High Content Screening32 platform. Having executed an HTS campaign, appropriate software is required to analyse the raw data that is generated to allow the quality control of all microtitre plates and identification of failed plates for repeat assays and this can be performed using the ActivityBase XE software suite. The result of an HTS campaign would be the construction of a primary hit list that would comprise compounds that appear to produce a suitable minimum effect upon the target. The activities of these compounds would need to be evaluated in confirmation assays and shown not to be an artefact such as optical interference or compound aggregation, thus suitable counter assays would need to be performed on the primary hits33-37. For cell based assays, inevitably, compounds that are cytotoxic will also exhibit profiles that are similar to inhibition. Once a compound hit list is prepared, they need to be assessed in a cytotoxicity assay e.g. Cell TitreGlo® from Promega that can be purchased in kit format and measures intracellular ATP38.

Figure 2 The industry standard assay development and High Throughput Screening infrastructure at the European ScreeningPort. The system contains (1) Hotel Workstation for microtitre plate handling and associated logistics, (2) Compound Workstation that stores microtitre plates containing compounds, (3) Opera Workstation for High Content Screening assays and (4) Reader Workstation that contains a microtitre plate washer and reader.
Pre-clinical drug discovery has relied substantially upon the development of assays that are subsequently utilised in high throughput campaigns to provide chemical starting points for drug discovery. Although these activities have produced a large number of candidate molecules, many have tended to fail in clinical trials due to their poor safety profiles and lack of efficacy. Initiatives such as industry-academic partnerships39,42, hypothesis driven screening43, virtual screening44, biomarker discovery45,46 and systems biology47,48 should all be employed in order to reduce the attrition currently observed in drug discovery.
References
1. Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006. 5. 993-996
2. Dixon SJ, Stockwell BR. Identifying druggable diseasemodifying gene products. Curr Opin Chem Biol. 2009. 13. 549-555
3. Giamas G, Man YL, Hirner H, Bischof J, Kramer K, Khan K, Ahmed SS, Stebbing J, Knippschild U. Kinases as targets in the treatment of solid tumors. Cell Signal. 2010. 22. 984-1002
4. Giacinti L, Vici P, Lopez M. Epigenome: a new target in cancer therapy. Clin Ter. 2008. 159. 347-360
5. Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins-novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov. 2008. 7. 841-853
6. Hormozdiari F, Salari R, Bafna V, Sahinalp SC. Proteinprotein interaction network evaluation for identifying potential drug targets. J Comput Biol. 2010. 17. 669-684
7. Posner BA. High-throughput screening-driven lead discovery: meeting the challenges of finding new therapeutics. Curr Opin Drug Discov Devel. 2005. 8. 487-494
8. Mayr LM, Bojanic D. Novel trends in high-throughput screening. Curr Opin Pharmacol. 2009. 9. 580-588
9. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nature Reviews Drug discovery. 2004. 3. 711-716
10. Kola I. The state of innovation in drug development. Clin Pharmacol Ther. 2008. 83. 227-230
11. Li X, Hu Y. Identifying the cellular targets of bioactive small molecules with activity-based probes. Curr Med Chem. 2010. 17. 3030-3044
12. Gribbon P, Sewing A. High-throughput drug discovery: what can we expect from HTS? Drug Discov Today. 2005. 10. 17-22
13. http://www.sbsonline.com/msdc/pdf/OPv2002-06-05.pdf; http://www.sbsonline.com/msdc/pdf/ANSI_SBS_4-2004.pdf; http://www.sbsonline.com/msdc/pdf/ANSI_SBS_3-2004.pdf; http://www.sbsonline.com/msdc/pdf/ANSI_SBS_2-2004.pdf; http://www.sbsonline.com/msdc/pdf/ANSI_SBS_1-2004.pdf.
14. Sharlow ER, Leimgruber S, Yellow-Duke A, Barrett R, Wang QJ, Lazo JS. Development, validation and implementation of immobilized metal affinity for phosphochemicals (IMAP)-based high-throughput screening assays for low-molecular-weight compound libraries. Nat Protoc. 2008. 3. 1350-1363
15. Carlson CB, Robers MB, Vogel KW, Machleidt T. Development of LanthaScreen cellular assays for key components within the PI3K/AKT/mTOR pathway. J Biomol Screen. 2009. 14. 121-132
16. Robers MB, Machleidt T, Carlson CB, Bi K. Cellular LanthaScreen and beta-lactamase reporter assays for high-throughput screening of JAK2 inhibitors. Assay Drug Dev Technol. 2008. 6. 519-529
17. Carlson CB, Mashock MJ, Bi K. BacMam-enabled LanthaScreen cellular assays for PI3K/Akt pathway compound profiling in disease-relevant cell backgrounds. J Biomol Screen. 2010. 15. 327-334
18. Zhang, J. H., Chung, T. D. and Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999. 4. 67-73
19. Rollinger JM, Langer T, Stuppner H. Strategies for efficient lead structure discovery from natural products. Curr Med Chem. 2006. 13. 1491-1507
20. Schopfer U, Engeloch C, Stanek J, Girod M, Schuffenhauer A, Jacoby E, Acklin P. The Novartis compound archive-from concept to reality. Comb Chem High Throughput Screen. 2005. 8. 513-519
21. Blaxill Z, Holland-Crimmin S, Lifely R. Stability through the ages: the GSK experience. J Biomol Screen. 2009. 14. 547-556
22. Quintero C, Kariv I. Design and implementation of an automated compound management system in support of lead optimization. J Biomol Screen. 2009. 14. 499-508
23. Johnson CW, Chatterjee M, Kubala S, Helm D, Houston J, Banks M. Efficient and effective compound management to support lead optimization. J Biomol Screen. 2009. 14. 523-530
24. Matson SL, Chatterjee M, Stock DA, Leet JE, Dumas EA, Ferrante CD, Monahan WE, Cook LS, Watson J, Cloutier NJ, Ferrante MA, Houston JG, Banks MN. Best practices in compound management for preserving compound integrity and accurately providing samples for assays. J Biomol Screen. 2009. 14. 476-484
25. Mayr LM, Bojanic D. Novel trends in high-throughput screening. Curr Opin Pharmacol. 2009. 9. 580-588
26. Colas P. High-throughput screening assays to discover small-molecule inhibitors of protein interactions. Curr Drug Discov Technol. 2008. 5. 190-199
27. Kumar RA, Clark DS. High-throughput screening of biocatalytic activity: applications in drug discovery. Curr Opin Chem Biol. 2006. 10. 162-168
28. Grant RJ, Roberts K, Pointon C, Hodgson C, Womersley L, Jones DC, Tang E. Achieving accurate compound concentration in cell-based screening: validation of acoustic droplet ejection technology. J Biomol Screen. 2009. 14. 452-459
29. Shultz S, Worzella T, Gallagher A, Shieh J, Goueli S, Hsiao K, Vidugiriene J. Miniaturized GPCR signaling studies in 1536-well format. J Biomol Tech. 2008. 19. 267-274
30. Varin T, Gubler H, Parker CN, Zhang JH, Raman P, Ertl P, Schuffenhauer A. Compound Set Enrichment: A Novel Approach to Analysis of Primary HTS Data. J Chem Inf Model. 2010. 50. 2067-2078
31. Mballo C, Makarenkov V. Using machine learning methods to predict experimental high-throughput screening data. Comb Chem High Throughput Screen. 2010. 13. 430-441
32. Zanella F, Lorens JB, Link W. High content screening: seeing is believing. Trends Biotechnol. 2010. 28. 237-245
33. Thorne N, Auld DS, Inglese J. Apparent activity in highthroughput screening: origins of compounddependent assay interference. Curr Opin Chem Biol. 2010. 14. 315-324.
34. Gribbon P, Sewing A. Fluorescence readouts in HTS: no gain without pain? Drug Discov Today. 2003. 8. 1035-1043
35. Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J Med Chem. 2010. 53. 37-51
36. Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007. 50. 2385-2390
37. Auld DS, Thorne N, Nguyen D-T, Inglese J. A Specific Mechanism for Nonspecific Activation in Reporter-Gene Assays. ACS Chem Biol. 2008. 3. 463-470
38. Petty WJ, Dragnev KH, Memoli VA, Ma Y, Desai NB, Biddle A, Davis TH, Nugent WC, Memoli N, Hamilton M, Iwata KK, Rigas JR, Dmitrovsky E. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin Cancer Res. 2004. 10. 7547-7554
39. Tralau-Stewart CJ, Wyatt CA, Kleyn DE, Ayad A. Drug discovery: new models for industry-academic partnerships. Drug Discov Today. 2009. 14. 95-101
40. Clark RL, Johnston BF, Mackay SP, Breslin CJ, Robertson MN, Harvey AL. The Drug discovery Portal: a resource to enhance drug discovery from academia. Drug Discov Today. 2010. 15. 679-683
41. Müller S, Weigelt J. Open-access public-private partnerships to enable drug discovery-new approaches. IDrugs. 2010. 13. 175-180
42. Roy A, McDonald PR, Sittampalam S, Chaguturu R. Open access high throughput drug discovery in the public domain: a mount everest in the making. Curr Pharm Biotechnol. 2010. 11. 764-778
43. Schopfer U, Engeloch C, Höhn F, Mees H, Leeds J, Glickman F, Scheel G, Ferrand S, Fekkes P, Pfeifer M. Hypothesis-driven screening. Methods Mol Biol. 2009. 575. 297-316
44. Schneider G. Virtual screening: an endless staircase? Nat Rev Drug Discov. 2010. 9. 273-276
45. van Gool AJ, Henry B, Sprengers ED. From biomarker strategies to biomarker activities and back. Drug Discov Today. 2010. 15. 121-126
46. Soares HD. The use of mechanistic biomarkers for evaluating investigational CNS compounds in early drug development. Curr Opin Investig Drugs. 2010. 11. 795-801
47. Schrattenholz A, Soskić V. What does systems biology mean for drug development? Curr Med Chem. 2008. 15. 1520-1528
48. Ho RL, Lieu CA. Systems biology: an evolving approach in drug discovery and development. Drugs R D. 2008. 9. 203-216
About the Author
Dr Sheraz Gul is Vice President and Head of Biology at European ScreeningPort, Hamburg, Germany. He is responsible for the management and development of Medium and High Throughput Screening activities for academic partners across Europe. He has 12 years research and development experience in both academia (University of London) and industry (GlaxoSmithKline Pharmaceuticals). This has ranged from the detailed study of catalysis by biological catalysts (enzymes and catalytic antibodies) to the design and development of assays for High Throughput Screening for the major biological target classes. He is the co-author of numerous papers, chapters and the Enzyme Assays: Essential Data handbook.
