

Modernising the supply chain using continuous manufacturing
Posted: 15 November 2017 | Dave Elder, Ronak Savla, Stephen Tindal | No comments yet
When the United States Food and Drug Administration (FDA) called for a shakeup of pharmaceutical production in 2002 it recommended the early adoption of technological advances and the application of modern quality management techniques. It believed that this would make manufacturing processes more robust and improve product quality.1

The European Medicines Agency (EMA) adopted a similar stance and updated its regulatory framework to support continuous manufacturing.2 The EMA recognised that continuous manufacturing dossiers are likely to be more complex, and recommended early dialogue with the agency, especially for legacy products transferred from batch to continuous manufacturing. EMA supports industry with scientific advice, process analytical technology (PAT) teams, subject matter expert (SME) office, and the Innovation Task Force (ITF).3
The FDA also recognised that adopting innovative manufacturing approaches could present significant challenges. There were concerns within the industry that adopting emerging technologies could result in delays to submissions. Therefore, the FDA initiated the Emerging Technology Team (ETT) to embed awareness into the agency4 and it has held over 15 ETT-Industry meetings since 2014. These meetings typically discuss API and drug product applications, as well as the control strategies that are being developed.5 These meetings are expected to achieve:
- Provide an overview of the proposed novel test/process/technology, including how it will modernise the manufacturing base
- Provide an overview of the development plan and any perceived technical or regulatory impediments, together with a timeline for the proposed submission.4
Success of continuous manufacturing
The first NDA approval using continuous manufacturing for a fixed-dose combination tablet product, Orkambi (lumacaftor/ivacaftor), was approved by the FDA and EMA in 2015.6 Prezista was the first supplemental NDA approval (2016) for changing from an established batch process to a continuous manufacturing process. For Prezista, continuous manufacturing reduced both resource/facility requirements (two rooms for continuous process versus seven rooms for the batch process) and production timelines (one day for continuous vs two weeks for batch).7
These product approvals have ensured increased interest from several other pharmaceutical companies. Pfizer announced the opening of a continuous manufacturing plant in Germany.8 Merck is also evaluating continuous manufacturing.9 However, Pfizer has highlighted that it cannot justify continuous granulation for high-volume products due to the lack of harmonisation between regulatory agencies, but it remains positive about using this technology for smaller volume products.10
Whenever continuous manufacturing is discussed, concerns over the concepts of ‘batch’ and ‘batch release’ are frequently raised. It is worth discussing the definitions of both batch and continuous manufacturing further.
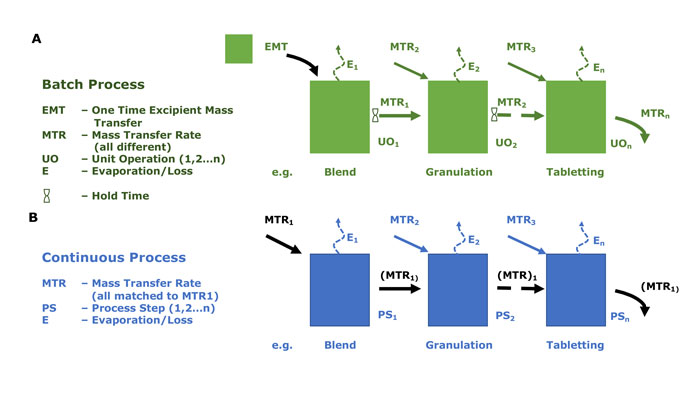
In batch processing, input materials, ie, API, water and excipients, are added to the system at the beginning of the process and the product is later discharged in its entirety, after proceeding through a series of unit operations (see Figure 1A). Intermediates are typically held for designated time periods before the full process (eg, powder blends, tablet cores, etc) is completed, as the unit operations often have different mass transfer rates. Some of the unit operations in the process could be considered as continuous, but the overall process is still completed in discreet batch modes. All material from one discreet batch is labelled with the same lot number.
In continuous processing, the input materials are continuously added and product discharged from the system, throughout the duration of the process (see Figure 1B). Thus, the output from each processing step moves continuously forward to the following processing step without a holding time. All process steps have the same rate of mass transfer and each must deliver material with suitable characteristics for input into the subsequent processing step, and the overall process runs at the rate of the slowest processing step. Labelling of material at the end necessarily involves some aspect of timestamping (instead of traditional batch numbering) to maintain traceability.
Continuous processing necessitates higher levels of process design, process monitoring and process control compared to those of standard batch processing.5 It can also offer opportunities to optimise and streamline batch processes that are sub-optimal. Thus, telescoping one-pot API synthesis, which is aligned with continuous flow chemistry, can circumvent the need for isolation/purification work-up stages in a classical batch process.11,12
Regulatory overview
Therefore, a ‘batch’ has ‘uniform character and quality within specified limits’. This is obviously closely linked to the product’s control strategy, which ensures that the process is always under control.13 The ‘batch’ definition also encompasses the total elapsed time of the production process, any variations encountered during production (ie, different lots of input material), the amount of product produced, and other parameters. One such important parameter includes ensuring appropriate traceability of all input materials at all times, allowing a complete reconstruction of the batch history. Some commentators have proposed a traceability resource unit (TRU).14 Traceability is particularly important in cases of product investigations, ie, out of specification (OOS), out of trend (OOT), consumer complaints or product recalls.5 There must be defined processes for start-ups and shut-downs, together with acceptance criteria for determining when product collection starts, or re-starts, as this is a significant part of the overall control strategy.
There are some differences in the regulatory expectations for continuous manufacturing, centred on the risk assessment process.15 These include:
- Any hazards are likely different from a standard batch process and the control strategies may also be different
- In the latter case, a model-based control system is frequently employed, involving multivariate monitoring, the analysis of very large data sets, elements of automation and PAT that can result in real-time release testing (RTRT)
- This, in turn, necessitates greater levels of interaction/communication between the various parties.
Process understanding
Quality by design (QbD)16 generates a body of knowledge (design space) through the use of design of experiments (DoE). Large amounts of process knowledge can be rapidly generated using continuous processing; however, the downside is increased usage of raw materials, particularly API.17 This increased knowledge can be augmented using statistically based process models. For example, sensitivity analysis can be used to identify key relationships/interactions between process parameters, allowing a multi-variant perspective of process understanding. These process models can, in turn, be used to develop control strategies for continuous manufacturing processes.18,19
In batch processes, scale-up implies either increasing the mass/equipment size ratio, or increasing the equipment size. Many of the biggest challenges in the batch processing in a QbD paradigm are related to scale up, including:
- Is the design space scalable?
- Are all the CPPs and CQAs scalable?
- Are there additional critical attributes observed after scaling?
In a continuous manufacturing paradigm, scale up is achieved by running the process for longer or faster, typically using smaller scale equipment. Thus, a 120,000 tablet/hour process, which is fully aligned with typical development scales, can produce one billion tablets/year – if run constantly 24/7 and 50+ weeks/year.17 Additional increases in product demand can be met by a) running the process longer, b) using additional capacity at the same scale – the so-called ‘parallel array’ approach, or c) increasing the process flow rate.13 Continuous manufacturing circumvents the need to change the equipment size, thus reducing costs and risks, by negating the need to supplement the additional design space with further batches at ‘commercial’ scale. Hot melt granulation/extrusion processes work on this principle. In a similar fashion, continuous primary processes, ie, drug substance, can be run continuously, but again at a smaller scale.20
Control strategy
The ability to identify, isolate and reject non-conforming material in real-time (or near real-time) is one of the key aspects of any continuous manufacturing control strategy.13 Thus, the evaluation and subsequent understanding of the reasons for such issues, the proportion of the batch affected and the justification for remedial measures are very important. As a consequence, a process control system needs to have at least the following features: a) rapid and controlled start-up and shutdown procedures (to minimise losses) b) continuous compliance of all CQAs by achieving steady-state conditions, and c) an assurance of quality irrespective of any system uncertainties or disturbance.21 This, in turn, necessitates various feedback, feed-forward control measures, etc, and the ability to trace OOT material or process inputs to a specified finished material. However, the linking of the on-line PAT monitoring system to the control system to facilitate this feedback/feed-forward control strategy still presents a significant challenge.
Real-time release testing
Real-time release testing (RTRT) is an integral part of the control strategy for a continuous process, as it allows for an active control system that can monitor the quality attributes of all incoming materials in real time using PAT. Process parameters can then be automatically modified to ensure conformance with the established acceptance criteria.13 RTRT can be applied during any stage of an API or drug product resulting in the exclusion of some, or indeed all, of the specified tests.22 That is, it is desirable to have so many on-line PAT measurements that off-line analysis is not fully required. However, the reality is that the sensitivity and selectivity of spectroscopically based PAT tools are not currently aligned with existing ICH Q3A23/Q3B24 impurity expectations.
The high frequency of data collection should result in a higher quality product, as a greater fraction of the batch (often 100-fold greater) has been sampled and analysed compared to a classical batch process.17 However, it also necessitates the application of statistical methodology for these larger sample sizes, which are required to enhance confidence that the batch conforms to the required quality.25 However, the use of ‘big data’ also necessitates changed the understanding of some historical concepts, such as uniformity of dosage units. Some pharmacopoeias, but not all, have introduced a revised approach to using large sample sizes for demonstrating content uniformity (Ph Eur 2.9.47).26 Nevertheless, the likelihood of observing OOS results is heightened from a statistical perspective when large volumes of data are generated. Additionally, sampling frequency needs to be aligned with on-line PAT capability. For example, many chemical reactions necessitate fast response times from the supporting PAT methodology, whereas most secondary processing – such as blending – would not.21
In addition, the real-time release also necessitates real-time storage of all relevant processing data and the challenges here are ‘data integrity’ considerations.27 Data integrity necessitates traceability of all facets of the product throughout the continuous processing stream and appropriate storage and retrieval of this processed data.21
Conclusion
Many of the manufacturing process steps that are required for continuous manufacturing already exist within many contract manufacturing organisations, eg, Catalent, where processes such as extrusion, roller compaction, tabletting and encapsulation are all recognised as continuous unit operations. Further consideration of the manufacturing, quality, regulatory and analytical/PAT/big data issues are required to develop the capabilities to support continuous manufacturing. Recent product launches have shown that both industry and regulators are willing to embrace continuous manufacturing in order to take advantage of key benefits that have already been demonstrated in other industries. In all likelihood, industry’s preferred strategy will be not to develop continuous manufacturing for NDAs in the short term, due to the costs and risk associated with tech transfers, and that it will be many years before even 50% of new products will be made using continuous manufacturing.
The initial investment necessary for continuous manufacturing is significant and includes novel equipment with limited prior knowledge. But in the long term, continuous manufacturing is more efficient and cost-effective than its batch counterpart. Data integrity is a current regulatory focus and the integrity of big data could present significant implementation challenges, but the overall patient benefit is likely to ensure that the development of continuous manufacturing progresses with increasing pace.
BIOGRAPHIES

Dave Elder has nearly 40 years of service within the pharmaceutical industry at Sterling, Syntext and GlaxoSmithKline. He is now an independent GMC consultant. Dr Elder is a visiting professor at King’s College, London, and is a member of the British Pharmacopoeia. He is a member of the Joint Pharmaceutical Analysis Group (JPAG), and a member of the Analytical Division Council of the Royal Society of Chemistry.

Stephen Tindal is Director, Science & Technology, at Catalent Pharma Solutions. He holds a Bachelor’s degree in Chemistry and Analytical Science from Loughborough University, UK. Stephen has worked at Catalent for 30 years, holding positions in R&D responsible for the formulation, process development, and clinical and commercial operations. He is part of Catalent’s Science and Technology Team and is based in Somerset, New Jersey, USA.

Ronak Savla is Manager, Scientific Affairs, Catalent Pharma Solutions. He received his PharmD and PhD degrees from Rutgers University. Ronak joined Catalent Pharma Solutions as a postdoctoral fellow in 2014 and transitioned to his current role as the Scientific Affairs Manager in June 2016. Ronak is a co-author of over 40 peer-reviewed papers, posters and articles.
References
- Pharmaceutical cGMPS for the 21st Century – A Risk-Based Approach: Second Progress Report and Implementation Plan, 21 August 2002.
- Hernan D. Continuous manufacturing: challenges and opportunities: EMA perspective. 3rd FDA/PQRI Conference on Advancing Product Quality, March 22-24, 2017.
- Mandate of the EMA Innovation Task Force (ITF). 6 August 2014, EMA/484400/2014, Human Medicines Research and Development Support Division.
- Advancement of emerging technology applications to modernize the pharmaceutical manufacturing base. Guidance for Industry. Centre for drug evaluation and research (CDER). December 2015.
- Lee SL, O’Connor TF, Yang X, Cruz CN, Chatterjee S, Madurawe RD, Moore CMV, Yu LX, Woodcock J. Modernizing Pharmaceutical Manufacturing: from Batch to Continuous Production. J Pharm Innov. 2015;10(3):191-199.
- Van Arnum P. Manufacturing trends in continuous mode. Pharm. Technol. 2015. http://connect.dcat.org/blogs/patricia-van-arnum/2015/09/18/manufacturing-trends-in-continuous-mode#.WWX18ITyvng. Accessed on 12 July 2017.
- Pharma Manufacturing. 2016. Advances in Pharmaceutical Supply Chain: Continuous Manufacturing (CM). https://www.pharmamanufacturing.com/assets/Media/2016/1604/Janssen-CM.pdf. Accessed on 29 July 2017.
- Healthcare Industry 07Jun2017. https://www.gesundheitsindustrie-bw.de/en/article/news/industry-40-pfizer-opens-continuous-manufacturing-plant-in-freiburg/. Accessed on 29 July.
- Glaad T. Merck’s Path To Continuous Manufacturing For Solid Oral Dose Products: What Stands In The Way? Life Science Leader, 8 December 2016. https://www.lifescienceleader.com/doc/merck-s-path-to-continuous-manufacturing-for-solid-oral-dose-products-what-stands-in-the-way-0001. Accessed on 29 July 2017.
- Stanton D. Pfizer: Global regulatory divergence restricted continuous manufacturing ambition. In-PharmaTechnologist. 29 June 2017. http://www.in-pharmatechnologist.com/Processing/Pfizer-on-continuous-manufacturing-and-regulatory-divergence. Accessed on 29 July 2017.
- Webb D, Jamison TF. 2010. Continuous flow multi-step organic synthesis. Chem Sci. 2010;1(6):675-680.
- Baxendale IR, Braatz RD, Hodnett BK, Jensen KF, Johnson MD, Sharratt P, Sherlock J-P, Florence AJ. Achieving continuous manufacturing: Technologies and approaches for synthesis, workup and isolation of drug substance May 20-21, 2014. Continuous manufacturing symposium. J Pharm Sci. 2015;104:781-791.
- Lee SL. Current FDA perspective for continuous manufacturing. MIT-CMAC 2nd International Symposium on Continuous Manufacturing of Pharmaceuticals, September 26-27, 2016.
- Moe T. Perspectives on traceability in food manufacture. Trends in Food Sci. Technol. 1998;9:211-214.
- ICH Q9. Quality Risk Management. Current step 4 version, 09 November 2005.
- ICH Q8(R2). Pharmaceutical development. Current step 4 version, August 2009.
- Byrn S, Futran M, Thomas H, Jayjock E, Maron N, Meyer RF, Myerson AS, Thien MP, Trout BP. Achieving continuous manufacturing for final dosage formation: Challenges and how to meet them May 20-21, 2014 Continuous manufacturing symposium. J Pharm Sci. 2015;104:792-802.
- Boukouvala F, Niotis V, Ramachandran R, Muzzio F. Ierapetritou MG. An integrated approach for dynamic flowsheet modelling and sensitivity analysis of a continuous tablet manufacturing process. Comput Chem Eng. 2012;42:30-47.
- Lakerveld R, Benyahia B, Braatz RD, Barton PI. Model-based design of a plant-wide control strategy for a continuous pharmaceutical plant. AIChE J. 2013;59:3671-3685.
- Cole KP, McClary Groh J, Johnson MD, Burcham CL, Campbell BM, Diseroad WD, Heller MR, Howell JR, Kallman NJ, Koenig TM, May SA, Miller RD, Mitchell D, Myers DP, Myers SS, Phillips JL, Polster CS, White TD, Cashman J, Hurley D, Moyla R, Sheehan P, Spencer RD, Desmond K, Desmond P, Gowran O. Kilogram-scale prexasertib monolactate monohydrate synthesis under continuous-flow CGMP conditions. Science 2017; 356:1144-1150.
- Rantanen J, Khinast J. The future of pharmaceutical manufacturing sciences. J Pharm Sci 2015;104:3612-3638.
- Elder DP. An overview of process analytical control. Eur Pharm Rev. 2017;20(6):44-47.
- ICH Q3A(R2). Impurities in new drug substances. Current step 4 version, 25 October 2006.
- ICHQ3B(R2). Impurities in new drug products. Current step 4 version, 02 June 2006.
- Schilling EG, Neubauer DV. Acceptance sampling in quality control. Boca Raton. 2009. 2nd Edition, CRC Press.
- Eur. 2.9.47. Demonstration of uniformity of dosage units using large sample sizes. Ph. Eur. 9.0: 384.
- Page T, Dubina H, Fillipi G, Guidat R, Patnaik S, Poechlauer P, Shering P, Guin M, Jonston C. Equipment and analytical companies meeting continuous challenges May 20-21, 2014 Continuous manufacturing symposium, J Pharm Sci. 2015;104: 821-831.
Issue
Related topics
Biopharmaceuticals, Bioprocessing, Bioproduction, Manufacturing





