Aqueous solubility-enhancing excipient technologies: a review of recent developments
Posted: 2 January 2018 | Henry Havel | No comments yet
At least some degree of solubility in water is necessary for active ingredients in pharmaceutical products to be effective in vivo. However, as efforts to discover and synthesise new active ingredients are pursued by industrial and academic medicinal chemists, achieving sufficient aqueous solubility can often be a significant limitation to clinical and commercial success…

Given the frequency and significance of this problem over the recent past, multiple solutions are being researched and applied in academia and industry.1 This review article provides a high-level description of the poor solubility problem and suggests a few techniques to deal with it.
The pharmaceutical discovery process can vary significantly depending on the characteristics of the intended target and therapeutic indication, availability of compounds with suitable properties to hit the target, and robustness of in vitro assays, among other differences. Irrespective of these differences, at some point in the discovery process the newly synthesised compounds will be dosed to animals to determine their pharmacological and toxicological activity in vivo. This is the point at which their aqueous solubility often becomes important. Occasionally, this realisation will occur during in vitro studies, but poor aqueous solubility is usually masked by the nature of these assays.2
Aqueous solubility is an obvious requirement if the chosen route of administration is intravenous or subcutaneous injection; aqueous vehicles are highly preferred (eg, as opposed to using solvents and surfactants) to minimise off-target effects due to the dosing vehicle. Additionally, it should be recognised that a solubilised formulation may precipitate in vivo and particles may be harmful because they can occlude capillary vessels. When the intended dose to the target animal cannot be solubilised in a typical aqueous vehicle, such as 5% dextrose or physiological saline, effort must be made to either apply a solubilising formulation to the existing compounds or direct the synthetic programme towards compounds with higher aqueous solubility.
It may be less obvious that compounds administered orally also need adequate aqueous solubility. For an oral medication, the solubility requirement stems from the fact that absorption typically occurs in the small intestine and only solubilised compounds can be absorbed. A simplified depiction of the oral absorption process is shown in Figure 1. The diagram also highlights the need for permeability through intestinal tissue. Since intestinal mucosa is principally hydrophobic, this characteristic usually means that hydrophobic compounds have better permeability than hydrophilic ones. Furthermore, successful oral absorption requires a balance of permeability and solubility. In recognition of these requirements, Pfizer researchers pioneered the ‘Rule of five’ concept in the late 1990s to direct medicinal chemists’ synthetic efforts.3 In a further elaboration of their approach, the concept of ‘Minimal acceptable solubility’ was developed, in which the importance of biological potency is incorporated,4,5 as illustrated in Figure 2.
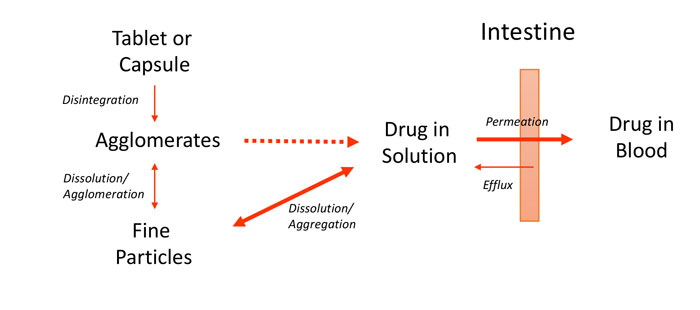
Figure 1: Simplified schematic of oral absorption process for solid dosage forms
While it should be noted that these predicted solubilities may not be accurate, what is important is the concept and trade-offs when considering solubility, permeability and potency for a series of discovery compounds to be administered orally…
These concepts have become more sophisticated over the past decade and researchers6,7 have incorporated them into the ‘Maximum absorbable dose’ metric to be used by pharmaceutical scientists8 as an important predictive tool during pharmaceutical discovery.
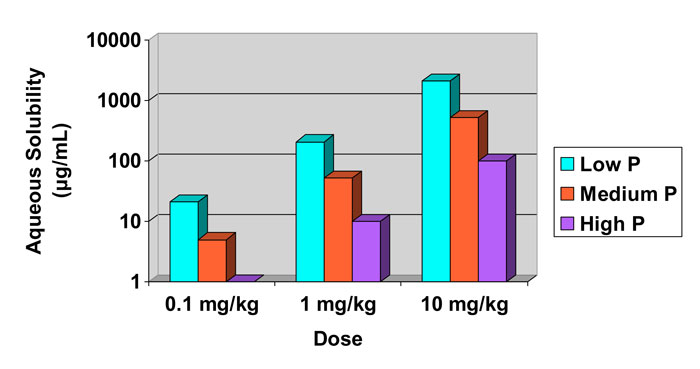
Figure 2: Calculated required aqueous solubility and its dependence on compound potency (in mg/kg) and intestinal permeability (P).4,5
Altering the chemical environment
When confronted with poor aqueous solubility for a parenteral product, it is relatively straightforward to improve solubility by altering the chemical environment of the compound. For instance, the use of buffers and counterions, along with pH adjustment, can be successful. This approach can be enhanced by incorporating pharmaceutically acceptable co-solvents (eg, ethanol, glycerol, polyethylene glycol, or propylene glycol), surfactants (eg, one of the polysorbates, a poloxamer or Cremophor) or cyclodextrins.9,10 The caveat to these solubilisation approaches, as has been noted previously, is that API precipitation in vivo often leads to irritation or even more severe reactions upon administration. A published review describes which solubilising excipients have been incorporated into commercial parenteral products up to 2004.11 A poorly soluble crystalline compound intended for administration by injection will often become sufficiently soluble if it is lyophilised to create an amorphous solid.12 Stabilising the amorphous form can be difficult, however, so this is not necessarily a panacea for poorly soluble compounds.13 Optimisation of the manufacturing process and the formulation (by incorporating stabilising excipients) is usually required to achieve sufficient physical and chemical stability for commercial products.
Solid form changes
Improving the aqueous solubility of orally administered compounds can often be accomplished by changing the solid form through various engineering technologies. For example, if the compound has an ionisable group then salt formation can often be explored. For compounds with or without ionisable groups, there has been the recent success in forming more soluble solid forms by creating a co-crystal.14,15 Both salt forms and co-crystals can be further optimised by conducting polymorph screening studies and selecting the most soluble polymorph.
Generating solid forms with small crystal sizes (eg, 2μm to 10μm through jet milling) will increase dissolution rate and systemic exposure for oral medications. Taking this particle size reduction approach even further, the use of nanocrystals (often between 0.2μm and 0.6μm) has proven advantageous for several poorly soluble compounds, resulting in five FDA-approved nanocrystal products for oral administration, most using the technology pioneered by Elan (now Alkermes).16
Because non-crystalline solids are generally more soluble than crystalline materials, oral formulations have been developed to take advantage of improved aqueous solubility of amorphous pharmaceuticals.12 The challenge with amorphous solids, however, is that crystallisation is preferred thermodynamically. Stabilised amorphous forms will be discussed in the next section, as excipients are required to achieve adequate physical stability.
Application of novel excipients
Although the conventional solubilising approaches illustrated thus far can improve aqueous solubility,11 many compounds remain challenging. Considerable effort has recently been devoted to identifying improved excipients and manufacturing processes for poorly water-soluble compounds. The two most commonly used categories will be discussed here as they are most commonly employed: lipid-based and amorphous-form stabilisers.
The use of lipids to deliver hydrophobic compounds through the intestinal lumen takes advantage of the ability of lipids to solubilise these compounds and that there are natural pathways to facilitate absorption of lipidic substances.17 Due to the myriad of lipid excipient choices, developing these formulations requires specialised knowledge and the necessity to screen a large number of excipient options before selecting the one combination that has the greatest chemical and physical stability. The product manufacturing process also requires unique equipment, as the liquid formulation is usually filled into soft gelatin capsules.
Recently, considerable effort has been devoted to the stabilisation of amorphous pharmaceuticals using excipients to trap compounds in physical configurations that have limited tendency to crystallise. Table 1 contains a list of some of these novel excipients. Foremost in this category are the polymers based on hydroxypropyl methylcellulose (HPMC)18 and its acetate succinate variation (HPMC AS).19 Pioneering work at Bend Research has shown how to create spray-dried dispersions with these and other polymers that stabilise many pharmaceuticals with poor aqueous solubility. As an alternative to spray-drying, melt extrusion has become quite popular and was the impetus for the polymeric excipient Soluplus20 and the polyvinyl alcohol product Parteck MXP.21 There has been recent progress in the quest to understand at the molecular level how these polymers work, and how to engineer even more effective polymeric solubilisers.22-24
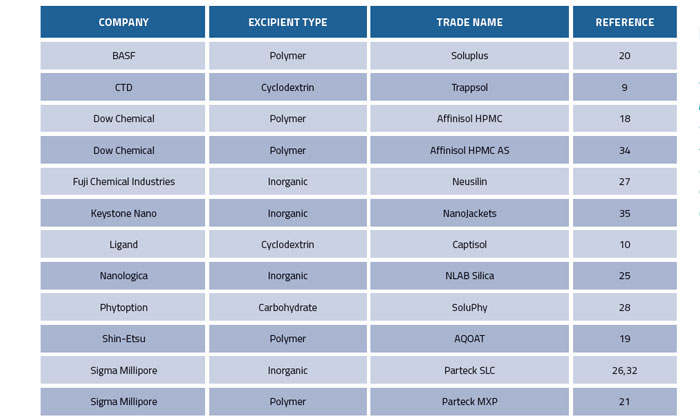
Table 1: Novel excipients used to solubilise pharmaceuticals
Mesoporous silica25,26 and other structured materials27,28 can solubilise APIs via an adsorption mechanism. However, to be effective as an oral product, the excipient must not only solubilise APIs but must also extend the time that the API stays in solution in vivo.29,30 Thus, researchers have employed the artificial stomach-duodenum model31 during formulation development to study prototype formulations and successfully explored combinations of polymeric and mesoporous silica excipients32,33 to enhance oral bioavailability of compounds with poor aqueous solubility.
References
- Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, Porter CJ. Strategies to address low drug solubility in discovery and development. Pharmacological reviews. 2013;65:315-499.
- Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and Prediction of Promiscuous Aggregating Inhibitors among Known Drugs. Journal of Medicinal Chemistry. 2003;46:4477-4486.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3-25.
- Curatolo W. Physical chemical properties of oral drug candidates in the discovery and exploratory development settings. Pharmaceutical Science & Technology Today. 1998;1:387-393.
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. Journal of Pharmacological and Toxicological Methods. 2000;44:235-249.
- Sun D, Yu LX, Hussain MA, Wall DA, Smith RL, Amidon GL. In vitro testing of drug absorption for drug ‘developability’ assessment: forming an interface between in vitro preclinical data and clinical outcome. Current opinion in drug discovery & development. 2004;7:75-85.
- Ding X, Rose JP, Van Gelder J. Developability assessment of clinical drug products with maximum absorbable doses. International Journal Of Pharmaceutics. 2012;427:260-269.
- Landis MS, Bhattachar S, Yazdanian M, Morrison J. Commentary: Why Pharmaceutical Scientists in Early Drug Discovery Are Critical for Influencing the Design and Selection of Optimal Drug Candidates. AAPS PharmSciTech. 2017. https://doi.org/10.1208/s12249-017-0849-3:1-10.
- Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. Journal Of Pharmaceutical Sciences. 1996;85:1017-1025.
- Kim Y, Oksanen DA, Massefski Jr W, Blake JF, Duffy EM, Chrunyk B. Inclusion complexation of ziprasidone mesylate with beta-cyclodextrin sulfobutyl ether. Journal Of Pharmaceutical Sciences. 1998; 87:1560-1567.
- Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharmaceutical Research. 2004;21:201-230.
- Hancock BC, Parks M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharmaceutical Research. 2000;17:397-404.
- Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Advanced drug delivery reviews. 2001;48:27-42.
- Blagden N, de Matas M, Gavan PT, York P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Advanced drug delivery reviews. 2007;59:617-630.
- Kuminek G, Cao F, Bahia de Oliveira da Rocha A, Goncalves Cardoso S, Rodriguez-Hornedo N. Cocrystals to facilitate delivery of poorly soluble compounds beyond-rule-of-5. Advanced drug delivery reviews. 2016;101:143-166.
- Liversidge GG, Cundy KC, Bishop JF, Czekai DA. 1992. Surface modified drug nanoparticles. U.S. Patent No. 5,145,684.
- Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Williams HD, Pouton CW, Charman WN, Bergstrom CAS, Porter CJH. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Advanced drug delivery reviews. 2016;101:167-194.
- Huang S, O’Donnell KP, Keen JM, Rickard MA, McGinity JW, Williams RO. A new extrudable form of hypromellose: AFFINISOL HPMC HME. AAPS PharmSciTech. 2016;17:106-119.
- Tanno F, Nishiyama Y, Kokubo H, Obara S. Evaluation of Hypromellose Acetate Succinate (HPMCAS) as a Carrier in Solid Dispersions. Drug Development and Industrial Pharmacy. 2004;30:9-17.
- Linn M, Collnot EM, Djuric D, Hempel K, Fabian E, Kolter K, Lehr CM. Soluplus as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. European journal of pharmaceutical sciences. 2012;45:336-343.
- Zheng M, Bauer F, Birk G, Lubda D. Polyvinyl alcohol in hot melt extrusion to improve the solubility of drugs. European Pharmaceutical Review. 2017;3. Scientific Poster Gallery.
- Mosquera-Giraldo LI, Borca CH, Meng X, Edgar KJ, Slipchenko LV, Taylor LS. Mechanistic Design of Chemically Diverse Polymers with Applications in Oral Drug Delivery. Biomacromolecules. 2016;17:3659-3671.
- Irwin JJ, Pottel J, Zou L, Wen H, Zuk S, Zhang X, Sterling T, Shoichet BK, Lionberger R, Giacomini KM. A Molecular Basis for Innovation in Drug Excipients. Clin Pharmacol Ther. 2017;101:320-323.
- Johnson LM, Li Z, LaBelle AJ, Bates FS, Lodge TP, Hillmyer MA. Impact of Polymer Excipient Molar Mass and End Groups on Hydrophobic Drug Solubility Enhancement. Macromolecules. 2017;50:1102-1112.
- Zhou C, Xia X, Garcia-Bennett AE. 2017. Super-saturating delivery vehicles for poorly water-soluble pharmaceutical and cosmetic active ingredients and suppression of crystallization of pharmaceutical active ingredients. U.S. Patent No. 9,757,456.
- Wieber A, Graband M, Witt V, Bauer F, Lubda D. Bioavailability Enhancement of Poorly Soluble Combination Preparations: A Case Study of Simultaneous Loading of Two APIs on Mesoporous Silica (Parteck SLC). AAPS Annual Meeting. 2015. p M1195.
Biography
 DR HENRY HAVEL is a pharmaceutical development scientist with more than 35 years’ experience working on projects from discovery to launch. He recently retired from his position as a Senior Research Fellow at Lilly and is now the Principal Investigator for Phytoption, a Purdue University start-up. Prior to joining Lilly, Dr Havel worked at The Upjohn Company (now Pfizer), Kalamazoo, Michigan. He has a PhD in Chemistry from the University of Minnesota.
DR HENRY HAVEL is a pharmaceutical development scientist with more than 35 years’ experience working on projects from discovery to launch. He recently retired from his position as a Senior Research Fellow at Lilly and is now the Principal Investigator for Phytoption, a Purdue University start-up. Prior to joining Lilly, Dr Havel worked at The Upjohn Company (now Pfizer), Kalamazoo, Michigan. He has a PhD in Chemistry from the University of Minnesota.
Issue
Related topics
Assays, Drug Discovery, Drug Targets, Polymorphism Screening