

Understanding the importance of diversity of particle size methods
Posted: 14 September 2018 | Edislav Lekšić (Almac Group) | 1 comment
The particle size of Active Pharmaceutical Ingredients (API) has a significant effect on a drug product’s manufacturability and performance. With respect to manufacturability, particle size can affect compatibility, flowability and blend uniformity; with respect to product performance it can affect solubility, dissolution, and bioavailability.
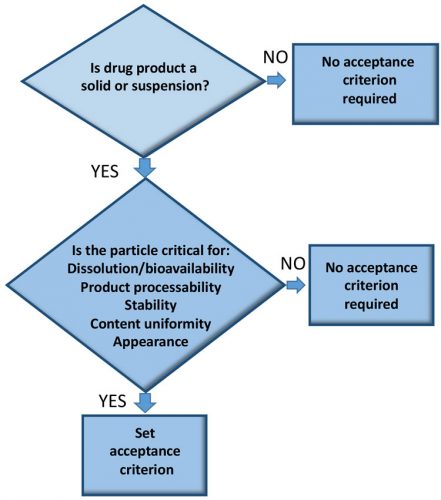
WHILE most manufacturers employ methods to control particle size prior to product release, particularly if the API is not soluble, they do not always consider the impact of particle size on product manufacturability (Figure 1).1 We recommend the monitoring of particle size at all manufacturing stages, as doing so will contribute to the understanding of, and ability to control, the process and ensure quality and consistency in batches. Different methods will be needed for monitoring particle size at different points in the process.
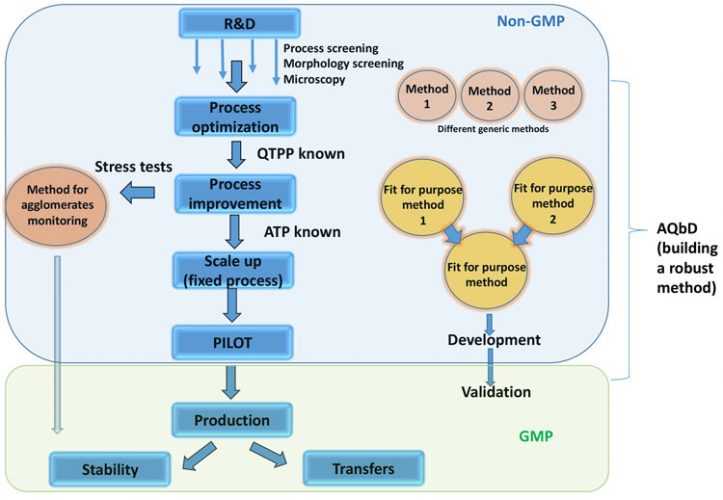
The decision of when to monitor particle size – and using what method – depends on how relevant the particle size is to the product. There is great diversity in the PSD (particle-size distribution) methods that can be used on the same material during drug product development, depending on what property is being measured. Programme leaders and manufacturers are often confused by the array of PSD methods, as there is no general PSD method listed in pharmacopoeias. This article explains the different PSD methods that are based on laser diffraction and clarifies their potential applications in different project phases and for different purposes (Figure 2).
The source of the diversity in PSD methods
The wide diversity of PSD methods stems from differences in their technical aspects (instrument parameters), their purpose (process monitoring at different project stages) and their relation to drug performance (primary particles, agglomerates).
The need for different method parameters is understandable.The common parameters that can vary in the wet dispersion method are pump speed, obscuration, and sonication time. There are different acceptable values for these parameters for smaller (eg, Dv90 less than 10µm) or larger particle sizes. While larger particles would require a higher pump speed to ensure efficient dispersion (preventing sedimentation inside the measuring cell), special care needs to be taken when setting the obscuration limits for samples characterised by small particle size to avoid phenomena such as multiple scattering.2,3
Should the method parameters change in a Good Manufacturing Practice (GMP) environment, it will most likely result in a new method that requires fresh validation – unless the method’s robustness was demonstrated under those parameters during the original validation. The idea of using one selected PSD method with the intention of covering a wide range of particle sizes – for example, where different API grades are targeted or when PSD methods support the micronisation of trials – presents a challenge.
There might be more than one PSD method suitable for determining the particle size of a given batch. For example, in the early stage of development, one might want to use a generic PSD method for monitoring the process. The term generic here refers to the method parameters that might be suitable, based on the particle size as observed under a microscope, eg, the default. By monitoring different samples during the early R&D phase using one or more generic methods, one can see trends in the changes in powder bulk properties and learn about the influence of process change on the particle size and morphology in general. Any difference in process (synthesis, scale up, and process optimisation) or improvement (such as reactor size, and increase or decrease in crystallisation temperature) might lead to differences in crystal growth or the formation of the agglomeration of different strength and sizes.
At a certain project stage, it is desirable to develop a fit-for-purpose method. Such methods are suitable for monitoring release or manufacturing processes, such as drying and milling. Often, the drying process is not properly monitored with respect to how the particle size and shape of those particles change during the course of the drying process. Thus, this is a production step that can potentially cause issues related to poor macroscopic property of the bulk. Milling is a critical step in the production of APIs because it significantly alters the physical property of the batch (eg, kinetics of solubility). It can also reduce the stability of the material because the increase in hygroscopicity, due to higher surface area or amorphous phase formation, can trigger secondary agglomeration formation upon storage.
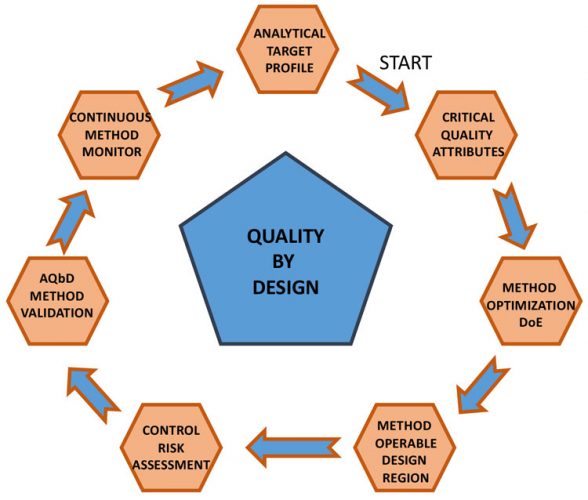
PSD methods are process related and, most likely, the method parameters should be fine-tuned to support PSD and morphology changes until the process for synthesis and isolation of a given API is fixed. This can then be followed by validation, depending on the project’s clinical phase. Continuously monitoring PSD and collecting data (which also involves microscopic analysis) is part of an Analytical Quality by Design (AQbD) approach to building a robust method that will be easy to validate when required (Figure 3).4
GMP methods
Release
The PSD method selected for use in the release should be in line with the Quality Target Product Profile(QTPP), which is part of a Quality by Design (QbD) approach to product development. The QTPP is a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality.4These product characteristics can be achieved by measuring primary particles or agglomerates in the sample. In most cases, due to low water solubility, PSD methods are used to measure primary particles (single crystals) in the bulk, as there is direct correlation between particle size and kinetics of solubility. In the case of crystalline material, it is to be expected that API consisting of smaller particles will show faster kinetics of solubility, which might influence bioavailability. Inhaled drug products comprised of fine and homogenous powders of primary particles characterised by narrow distribution are important for successful particle deposition and product efficiency. In most cases, production batches consist of a mixture of primary particles and agglomerates.Agglomerates are objects in which more particles are joined together and act as one. Their degree of hardness is often different, which increases the sample’s heterogeneity. Further discussion is required to understand formulation type as the presence of agglomerates might have a significant role in terms of solubility, formulation performance, and content uniformity. In such cases, measuring the agglomerates might be desirable. Agglomerates formation might be created intentionally to improve the process, such as through an increase in filterability time.
Stability
In general, treating the PSD method as a default quality test in an official GMP stability study should be an exception rather than a rule. There are two main reasons for this:
- Any resulting increase in particle size does not necessarily reflect a quality issue
- The PSD method might not be feasible after the powder macroscopic property has been changed.
The main purpose of the stability study is to determine the shelf–life of drug substances and drug products by stressing the samples. However, changing the macroscopic property of the powder under stability conditions, such as particle size via agglomeration formation, might not be relevant for the quality aspects of a given compound. Out-of-precision (OOP) results could occur due to sample inhomogeneity and the small sample quantity used for each stability point testing.5 Out-of-specification (OOS) or out-of-trend (OOT) results found by executing the stability protocol might not have any meaningful influence on the quality of the product. For instance, weak agglomeration formation that can be detected with a suitable PSD method during stability testing will generate results that will trigger an OOS investigation in a GMP environment. The results could be misleading as this change might not be relevant for the product quality, given that formed agglomerates will be destroyed by wet granulation.
In addition, stress conditions created by temperature and humidity changes can significantly alter the macroscopic properties in the bulk, such as powder flow and powder cohesiveness. An existing (eg, dry dispersion) method may no longer be suitable because the method’s measurement parameters were optimised during method development on the batches of the same compound, but with different macroscopic properties. This indicates the need to have different PSD stability methods in line with the QTPP, which may differ from the release specifications (for instance, omitting the sonication step during sample preparation). Changing the method to adjust the parameters in a GMP environment is not easy to justify, causes lengthy delays in reporting the results, and might be quite expensive for manufacturers. Instead, the influence of agglomerate formation during storage and its impact on drug product performance should be examined in the early stages of drug development, and sensitive methods should be developed accordingly.
Batch-to-batch comparison methods
When a batch is comprised of agglomerates of different strengths, or when a batch is characterised by fragile crystals with poorly expressed crystal habit, methods that support batch-to-batch comparisons might be suitable. When agglomerates of different strengths are present in the bulk, they cause material to be heterogeneous and therefore challenging to sample homogenously. Fragile crystals break randomly, making it difficult to achieve a plateau during sonication, which is needed to define full sample dispersion. Batch-to-batch comparisons do not provide real particle size (eg,primary particles), but particles formed upon application of certain method conditions (eg, stress caused by sonication) serve to differentiate among batches.
Conclusion
During the project development more than one PSD method might be employed as PSD methods are sensitive to chemical process changes. As a final release test, a PSD method is selected to ensure consistency and quality directly related to known product performances, while PSD results obtained during a stability programme contribute to the determination of a drug product’s shelf life.
REFERENCES
- ICH Guideline Q6A, Specifications: Test procedures and acceptance criteria for new drug substances and drug products: Chemical Substances, 2000.
- Malvern Instruments Limited, Application note MRK1902-02, Wet or liquid dispersion method development for laser diffraction particle size measurements, UK, 2013.
- Malvern Instruments Limited, Technical note TN130222, Dry powder method development for laser diffraction particle size distribution measurements, UK, 2015.
- ICH Guideline Q8(R2) Pharmaceutical development. Technical Requirements for Registration of pharmaceuticals for Human Use Geneva, Switzerland, 2009.
- a) ISO13320, 2009. Particle Size Analysis – Laser Diffraction Methods, Part 1, General Principles, b) USP29 <429> Light Diffraction Measurements of Particle Size, c) EP8.0, 2.9.31. Particle Size Analysis by Laser Light Diffraction.
BIOGRAPHY
DR EDISLAV Lekšić is the Team Leader of Physical Sciences at Almac Sciences. He has worked in the pharmaceutical industry for the past 12 years, with expertise in solid state chemistry related to API target selection, crystal surface property, scale-up and production troubleshooting. He has been involved in preformulation work and development of complex systems such as solid dispersions and cocrystals. For several years he has worked closely with patent law authorities contributing to aspects of IP. He has a strong GMP background and experience related to XRPD and PSD method validation and transfers. Edislav received a PhD in supramolecular chemistry from the University of Zagreb.
The rest of this content is restricted - login or subscribe free to access
 Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Why subscribe? Join our growing community of thousands of industry professionals and gain access to:
- bi-monthly issues in print and/or digital format
- case studies, whitepapers, webinars and industry-leading content
- breaking news and features
- our extensive online archive of thousands of articles and years of past issues
- ...And it's all free!
Click here to Subscribe today Login here





psd description is interesting in guideline through ich,