

It’s not just about the science: microbiological analysis
Posted: 3 May 2019 | Alice Laures - GSK, Lisa Wysocki - GSK, Paul J Newby - GSK | No comments yet
Modernisation of microbiological test methods involves far more than just the science. As new systems and technologies become available it becomes increasingly important that potential users can identify not only the technical requirements, but also the business case. Here, Paul Newby, Alice Laures and Lisa Wysocki from GlaxoSmithKline discuss criteria they have developed to assist in the selection process during consideration of new microbiological environmental monitoring technologies.
Pharmaceutical microbiology is an applied branch of microbiology, which involves the study of microorganisms associated with the manufacture of pharmaceuticals. During manufacture, minimising the number of microorganisms in a process environment, excluding microorganisms and microbial biproducts like exotoxin and endotoxin from water and other starting materials, and ensuring the finished pharmaceutical product meets it’s registered microbiological release specification are all essential elements of any microbiological control strategy.2,6,15,18,19 Drug product safety is a major focus of pharmaceutical microbiology. Microbiological risk is related to the intended patient group and route of administration. Sterile parenteral products being highest potential risk to patient safety and oral solid dosage forms among the lowest.6,16,18,19 Microbiological control of drug products is controlled to a level consistent with patient safety. A pragmatic approach to microbiological contamination control is required which includes input material bioburden and environmental monitoring of manufacture facilities.19 Pathogenic bacteria, fungi (yeasts and moulds) and toxins produced by microorganisms are all possible contaminants of medicines- although stringent, regulated processes are in place to ensure the risk is minimal.
Microbiological environmental monitoring (EM) is an essential element for microbiological control and a mandatory requirement for pharmaceutical manufacturing facilities.6,2 The data generated and the information provided via trending, are essential elements of the microbiological control strategy for drug product manufacture.1,17 EM consists of air, surface and may include operator monitoring, depending on the dosage form and intended patient group. Wider facility monitoring will also include microbiological water analysis, for both water system monitoring and as an input raw material ingredient (Figure 1).
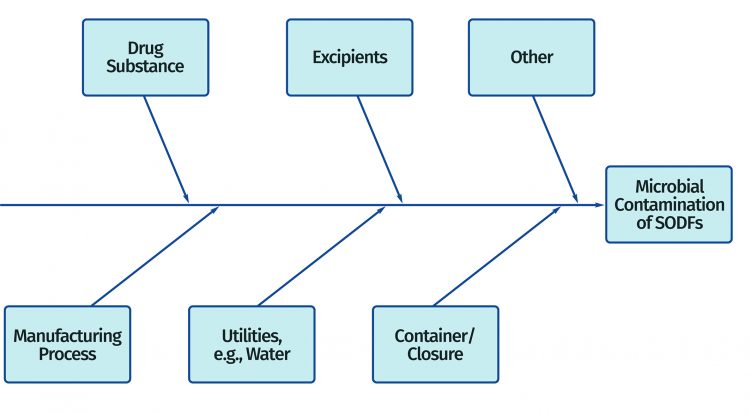
Figure 1: Fishbone diagram identifying potential sources of microbial contamination for a packaged solid oral dosage form (SODF)
The schematic in Figure 1 is for nonsterile Solid Oral Dosage Forms (SODF) however, the approach outlined is equally applicable to other nonsterile and sterile dosage forms. Action and alert limits are developed to reflect regulatory guidance and facility performance.1 The data generated during EM are compared to the relevant action and alert limits to demonstrate microbiological control of production facilities, or in the case of action /alert breaches where control may be starting to or has been lost.
Current microbiological methods for EM include settle plates, active air monitoring, swab and contact plates for surfaces and operator testing, as well as membrane filtration for water. These conventional approaches are commonly growth-based methods. Microorganisms isolated by any of these methods must be incubated and grown as individual colonies to obtain a result. Time to result for these growth-methods take several days. Additional time is needed if microbial identification is required. Real-time data for air, surface and water quality and operator monitoring is the desired state, but is not possible with current methods.7,8,9,10 Real-time data would be a significant advantage and would enable timely intervention, troubleshooting and remedial actions to avoid loss of microbiological control of manufacturing environments. This quicker time to result would ultimately prevent or reduce the potential for product contamination and associated cost. Evolving rapid alternatives to conventional EM methods offer the possibility of quicker time to result, integrated data integrity functionality and increases in productivity.
There are significant challenges, both technical and financial, associated with the introduction of new microbiology test methods in the pharmaceutical industry so why go through the efforts involved to change?7,8 What are the drivers? Is there a real need, or is this simply change for change’s sake? After all, current methods have been perfectly sound for decades, and in fact, continue to satisfy current regulatory and practical test requirements for many conventional small molecule chemical entities.
The short answer is simply that the drive towards large molecules, cell and gene therapies and advanced methods of manufacture are a step change away from the industry of the past and require quicker, more productive, higher throughput and data integrity compliant product analysis solutions. This includes pharmaceutical microbiology which must modernise in the face of new business realities.
The pharmaceutical industry today is facing a period of unprecedented change. Pressure is coming from many areas, including world-wide restructuring of healthcare systems, personalised medicines, data integrity functionality, high manufacture throughput with increased customer demand for cheaper, more effective therapies and increased competition from generic manufacturers. Technological innovation has an important part to play in the search for solutions to many of these issues.
There is a real and growing need in pharmaceutical microbiology for the introduction of new analytical methods which can address the requirements of today’s fast-paced industry. Current conventional growth-based microbiological techniques date from the nineteenth century. Technology driven solutions to drug development and manufacture are beginning to take shape. Initiatives such as the FDA’s Process Analytical Technology, the publication of pharmacopoeial chapters such as the USP chapter 1223, European Pharmacopoeia chapter 5.1.6 and guidance documents such as the PDA technical report 33, are helping to outline the technical and validation requirements necessary for new microbiological test technologies.
The emergence of cell-based therapies, biopharmaceutical drug products and new continuous processes will all benefit from real-time or near real-time analytical data and very different types of analytical evaluation. But the rate of uptake of alternative microbiological methods remains slow.
Regulatory agencies also support pharmaceutical innovation and modernisation. The agencies recognise that the adoption of innovative approaches may represent challenges to industry, but support modernisation and improving quality. The FDA has an Emerging Technology Team, ETT, to which companies can submit questions and proposals about the use of specific emerging technology. The Medicines and Healthcare Products Regulatory Agency (MHRA) has launched an ‘Innovation Office’ to help organisations who are developing innovative medicines, medical devices or using novel manufacturing processes to navigate the regulatory processes in order to be able to progress their products or technologies. It is also significant that companies are now working together in the pre-competitive space to support the modernisation of microbiological test methods. The Microbiology Modernisation Cross-industry Consortium (MMCC), a cross industry consortium, has recently been formed between a number of pharma companies including AstraZeneca, Merck, GSK, Pfizer, J&J, Janssen and Sanofi to help build the case for new microbiological technology uptake ( contact authors for further details).
Building the case for environmental monitoring (EM)
The selection process for the right technology platform is an important part of the overall move towards modernising microbiological methods. Selection is not a simple task due to the complexity of many test platforms and the lack of familiarity of the prospective purchaser with the technology involved. To help in the assessment stage when considering the range of technologies several key requirements have been developed for the following areas:
- technical criteria
- data integrity & archiving
- business criteria
The purpose of these criteria is to aid prospective customers in their initial assessment of new technology, in gathering key information from suppliers to make an informed assessment to determine if the technology will be suitable for their needs. The criteria fall outside of the system qualification phase and are not intended as a substitute for a User Requirement Specification (URS) as outlined in USP 1223. If individual criteria are not met, or suppliers are unable to provide sufficient justification, it is important for the prospective customer to consider the significance of the unmet criteria in relation to suitability of the system and the other criteria that do meet requirements. It is important to highlight that these requirements are designed as far as possible, to be technology agnostic. They can be used to engage with vendors. They are also intended to be used during the seeking phase of a new technology to form the basis of an evaluation protocol as well as assisting with an evaluation go/no go decision. See Figure 2.
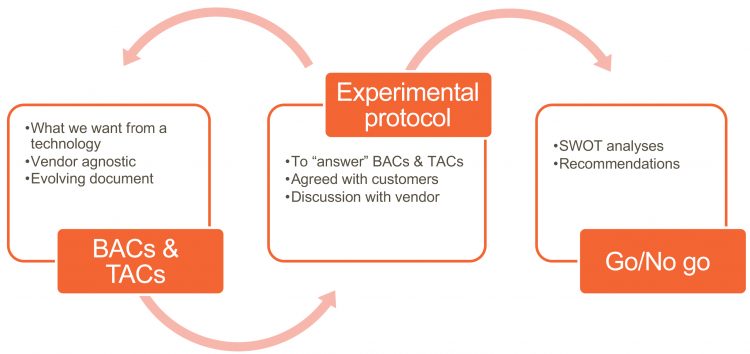
Figure 2: Process flow for use of BACs and TACs in technology selection and evaluation
Definitions
The definitions below are considered critical to the correct understanding of this document.
Term definition
Must: A first intent requirement
Should: A recommendation or suggestion
May: Information to aid understanding, as guidance or as an example
Technical criteria
This section details technical acceptance criteria. They represent points to be raised by a potential customer with the supplier of any new technology-based EM test system.
1.1 Performance equivalence* of the system must be equivalent to or better than the compendial methodology in terms of accuracy, precision, specificity, limit of detection, limit of quantitation, linearity, range and rates of false positive and negative results (reference USP 1223).
1.1.1 Equivalent to or better than the compendial methodology in terms of accuracy*.
1.1.2 Equivalent to or better than the compendial methodology in terms of precision*.
1.1.3 Equivalent to or better than the compendial methodology in terms of specificity*.
1.1.4 Equivalent to or better than the compendial methodology in terms of limit of detection*.
1.1.5 Equivalent to or better than the compendial methodology in terms of limit of quantitation*.
1.1.6 Equivalent to or better than the compendial methodology in terms of linearity*.
1.1.7 Equivalent to or better than the compendial methodology in terms of rates of false positive and negative results*.
1.2 The plate reader must have the ability to accurately read different media surfaces (rough/smooth/indented).
1.3 The system must enumerate and record the number of colonies present on plates.
*Refer to appendix 1
1.4 The system must be capable of recording and retaining an image of any growth present on plates.
1.5 The system must record numerical results for both positive and nil plates.
1.6 The system must be able to distinguish between viable and non-viable microorganisms and artefacts.
1.7 The system must have an incorporated mechanism for segregation of positive and nil plates following reading.
1.8 In the event of a failed result the system must allow further investigation.
1.9 The system must be applicable for sterile and non-sterile environmental samples (Grade A to D).
1.10 All product contact system consumables must be provided as sterile.
1.11 The system must incorporate controls to prevent contamination risk
1.12 The system and consumables must be compatible with commonly used site disinfectants and sterilants (eg, hypochlorite, H2O2).
1.13 The system must be LAN compatible.
1.14 The system footprint must be defined and compatible with the laboratory.
1.15 The system consumables must be compatible with plate bagging and the use of RABS (reference Appendix II) and sterility isolators.
1.16 Unique sample identifier.
1.16.1 Unique plate identifier must not obscure the reading mechanism.
1.16.2 Unique plate identifier must be applied to plate prior to sterilisation.
1.16.3 Unique plate identifiers must be resilient (not susceptible to decomposition).
1.16.4 Unique plate identifiers should be appended to a monitoring location (fixed or mobile), sampling time, duration, incubation status. Where applicable the system should be able to handle and read the following plates:
1.17.1 lockable and non-lockable plates
1.17.2 contact plates
1.17.3 membrane filtration plates
1.17.4 plates of different sizes, specifically 45mm, 60mm and 90mm
1.18 The system should provide the status of the plate and whether it is fit for use (eg, Certificate of Analysis, in house fertility test, expiry).
1.19 The system should be capable of interfacing with site batch release systems to record additional details such as the media type, media batch number, sample information …
1.20 The system should be capable of interfacing with LIMS systems in use
1.21 The system should include a failsafe feature to ensure correct incubation periods.
1.22 The system should be capable of tracking individual samples
1.23 The supplier should provide evidence of system robustness* and ruggedness*.
1.24 The system should include automation of loading, reading and sorting of plates to facilitate the loading of multiple plates at a given time.
1.25 The system may include a failsafe feature to ensure media has been first QC accepted.
1.26 The system may include a failsafe feature to ensure media has not expired.
1.27 The system may not open the plates to perform the analysis.
1.28 The system may include an incubator.
1.29 If the system includes an incubator, it must be able to incubate plates at an accuracy of +/-0.5 °C.
2. Data Integrity & archiving
This section details data integrity and archiving acceptance criteria (5,13). Like the technical acceptance criteria, they represent points to be raised by a potential customer with the supplier of any new technology-based EM test system.
2.1 Samples must be trackable via a unique sample identifier for the duration of the analysis.
2.2 The system must comply with FDA 21CFR Part 11.
2.3 The system must be able to capture and archive all data generated in accordance with data retention requirements (5,13))
2.4 Individual sample data must be retrievable.
2.5 The system must ensure that stored data or methods cannot be altered without changes being captured in a secure, time stamped and attributable audit trail.
2.6 The system must include capability for data back-up and recovery to remote (server based) location.
2.7 The system must have individual log-in security, which can attribute all actions within the application to specific individuals.
2.8 The system must be able to attribute distinct permissions to at least 2 User Groups (i.e. User and Administrator) and assign users of the system to the relevant user group
2.9 If users have a dual role, they must have a distinct user ID for each role including vendor and/or engineer access.
2.10 The system must have an audit trail that records both application access and all actions performed by the administrator and all users of the system.
2.11 Any data files, audit trails and method files must be protected from deletion within the system software
2.12 Any data files, audit trails and method files must be protected from deletion/modification outside of the application i.e. Windows Explorer.
2.13 The program name, current version number, original author, Date created, and modification history must be recorded within the source code. This should be recorded.
2.14 The system must be compatible with the Windows 10 operating system.
2.15 Unique sample identifiers should include, time stamp, duration, incubation status and sample set.
3 Business Criteria
New technologies often involve initial high purchase and operating costs. Business benefits and return on investment can be obscured with simple direct comparison of like for like with conventional methods. However, a broader evaluation looking at productivity gains, automation potential and higher throughput can present a very different picture. This section documents the business acceptance criteria to be considered when selecting an environmental monitoring system.
3.1 Efficiencies in the context of site demand and sample throughput
3.1.1 The system capacity must be defined and should be able to accommodate a minimum of 100 plates at one time.
3.1.2 The supplier should work with the customer to provide evidence of net time savings (eg, start-up time saving, total number of manufacturing and testing days, documentation & approval, sample preparation/movement).
3.1.3 The supplier should provide evidence that the methodology is equivalent or better than the compendial methodology in terms of simplicity (ie, number of consumables, procedure).
3.1.4 The supplier should provide evidence of error and investigation cost savings.
3.1.5 The level of competency required to operate the system should not exceed that of current QA scientists or require specialist knowledge.
3.1.6 The supplier should work with the customer to provide evidence of man hour savings.
3.2 Cost
3.2.1 The supplier must provide evidence of return on investment in terms of cost per analysis and cost per year in comparison to conventional test and capital, facilities and servicing costs over a 3-year period.
3.2.2 The supplier must be able to provide scalable pricing.
3.2.3 The cost and level of the service contract must be provided and itemised.
3.2.4 Any necessary modifications or changes to current laboratory testing environment should be fully explained and costed.
3.2.5 The upfront investment in terms of capital & facilities should be defined.
3.3 Acceptability of adoption and industry uptake
3.3.1 The supplier must provide evidence of local regulatory familiarisation.
3.3.2 Supported evidence in the scientific literature should be provided.
3.3.3 Evidence of wider uptake across industry should be provided.
3.4 Business risk in implementation
3.4.1 Shall this be necessary; the system methodology and operating procedures must allow revision to the previous ways of testing; for example, in the case of equipment breakdown.
3.4.2 The supplier must confirm that routine maintenance and repair should not be necessary more than twice a year.
3.4.3 The supplier must confirm that routine instrument downtime is no longer than 48 hours.
3.4.4 In case of instrument failure, the supplier must respond within a time frame of 24 hours.
3.4.5 If the instrument or critical consumables are unavailable, the supplier must provide evidence of a contingency plan.
3.4.6 The supplier must provide evidence of a disaster recovery plan.
3.4.7 The supplier must provide evidence that the supply of consumables can consistently meet Quality Assurance and Control requirements.
3.4.8 The system design must avoid negative impact on facility parameters, eg temperature, humidity, particulate levels.
3.4.9 The supplier must provide a lifecycle management plan for the equipment, which includes firmware, software and hardware/consumables upgrades. The upgrade processes, timelines and costs must be provided.
3.4.10 Multi-sources of non-proprietary consumables should be available.
Conclusions
Current microbiological EM test systems will be unsuitable for many of the new drug modality applications as they are too slow and labour intensive and not capable of delivering real-time or near real-time results. Speed to result is increasingly important for high cost and short duration manufacturing processes. Patient safety and product quality considerations for some advanced therapeutic drug products require a move towards real time or near real time EM analysis.
An additional driver for new approaches to microbiological analysis is that of data integrity. Guidance from authorities in recent years have underlined the importunate of data integrity principles in the control of analytical data. New technologies offer the potential for integrated solutions which will meet these emerging data integrity requirements. (Ref MHRA guidance, FDA GUIDANCE). It is no longer appropriate in a technology-driven industry of the twenty first century to use nineteenth century microbiological test methodologies requiring procedural workarounds to meet data integrity requirements.
New Rapid Microbiological Methods (RMMs) are emerging, which offer the potential to provide:
- Integrated data integrity functionality
- rapid results
- real or near real-time data
- in-line or at-line testing
- automation and high-test throughput potential
- high labour efficiency.
The stage is set for the implementation of a whole new generation of test systems in pharmaceutical microbiology including environmental monitoring evaluation.
Acknowledgements
The help and assistance of Julian Kay and David George in the preparation of the business and data integrity sections of this publication is greatly appreciated.
Biographies
Paul J Newby has worked in the field of pharmaceutical microbiology, in both research and manufacture roles, for the past 30 years. His current role is Microbiology Modernisation Lead in the Future Analytical and Control Technology group within GSK Research and Development. Paul is a former member of the BP expert committee on microbiology and was involved in reviewing the initial EP chapter on Rapid Methods (5.1.4). He was also part of the PDA technical working party charged with updating Technical Report 33 ‘Evaluation, Validation and Implementation of New Microbiological Testing Methods’. He has recently become a Fellow of the Royal Society of Biology.
Lisa Wysocki is a Microbiology Technical Lead and Associate Fellow at GlaxoSmithKline in the CMC Analytical Group. She provides microbiology support for Pharmaceutical Product Development and Supply and leads an effort on new technology development and implementation for rapid microbiology methods. She has previous experience in molecular biology, microbial genetics, mammalian cell culture and biological sample management.
Alice Laures works for GlaxoSmithKline R&D in the UK Medicines Research Centre where she leads the Future Analytical and Control Technologies group – a multi-disciplinary team of technical experts in mass spectrometry, novel imaging technologies and rapid microbiological techniques. Alice is a cofounder of the Microbiology Modernisation Cross-Industry Consortium (MMCC), set up in 2018 to create a collaborative forum for pharmaceutical companies interested in developing the use of novel microbiological technologies. Alice also has experience in pharmaceutical analysis and spectroscopy.
References
1. Rapid Microbiological Methods in the Pharmaceutical Industry, Easter, M.C., ed. Interpharm Press, 2003.
2. USP. General chapter <1223> Validation of Alternative Microbiological Methods. United States Pharmacopeial Convention.
3. Pharma Eur. Chapter 5.1.6 Alternative Methods for Control of Microbiological Quality. European Pharmacopoeia, European Directorate for the Quality of Medicines & HealthCare.
4. PDA Technical Report 33 Evaluation, Validation and Implementation of Alternative and Rapid Microbiological Methods. 2013 Parenteral Drug Association, Inc.
5. MHRA Rules & Guidance for Pharmaceutical Manufacturers and Distributors, 2017.
6. Elder D, Newby PJ. Risk-based Microbiological Testing, European Pharmaceutical Review 2016; 21(3): 36-40
7. Newby PJ. Implementation, Validation and Registration of Rapid Microbiological Methods, American Pharmaceutical Review 2004; 7(4), 10-15.
8. Paul M, Hooper S. One Company’s Approach to Rapid Micro Methods. PDA Letters, Rapid Microbiological Methods, October 2017.
9. PDA Technical Report No. 67, Exclusion of Objectionable Organisms from Non-Sterile Pharmaceuticals, Medical Devices and Cosmetics, 2014 Parenteral Drug Association, Inc.
10. MHRA GMP Data Integrity Guidance for Industry 2018.
11. PDA Technical Report 80, Data Integrity Management System for Pharmaceutical Laboratories, 2018 Parenteral Drug Association, Inc.
12. FDA (2004) Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice. <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070342.pdf>. (Accessed on 10 May 2017).





