Lead discovery for targeting G protein-coupled receptors
Posted: 19 October 2011 |
G protein-coupled receptors (GPCRs) control a plethora of key physiological functions in every cell of an organism. GPCRs are therefore involved in many diseases, since altered ligand or receptor levels and genetic or epigenetic modifications can lead to GPCR dysfunction and hence a pathophysiological phenotype. About one third of currently marketed drugs target GPCRs. The human genome contains 720-800 predicted GPCRs, and about half of them respond to olfactory/sensory signals, whereas the others are known or predicted to be activated by endogenous ligands and many of these represent potential drug targets. Seventy seven per cent of these non-sensory GPCRs belong to the class A (rhodopsin-like) family, whereas 14 per cent represent class B (secretin-like) GPCRs, less than one per cent belong to the class C (metabotropic receptor-like) or the atypical frizzled-/smoothened receptor class, and the remaining 25 per cent are orphan receptors…
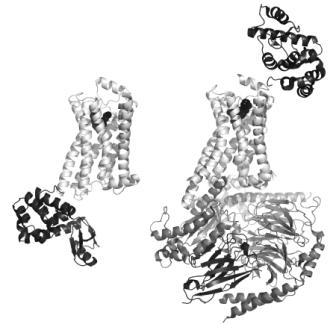
FIGURE 1 Structures of active and inactive conformations of the β2 adrenoceptor Structures of the β2 adrenoceptor (light grey) in an inactive conformation with the antagonist carazolol bound (left33), and in an active conformation in complex with Gs (dark grey) and the agonist BI-167107 bound (right41). In both cases, the ligand is represented by black spheres to show the location of the orthosteric binding site within the membrane-spanning region of the protein. T4 lysozyme (black) is inserted in the third intracellular loop between helices 5 and 6 in the inactive structure (bottom left), and fused in the extracellular N-terminus for the active structure (top right), to remove flexibility and to provide polar surfaces for crystallisation. The nanobody (black, bottom right) served to stabilise the open conformation of Gs and also provided crystallisation contacts
G protein-coupled receptors (GPCRs) control a plethora of key physiological functions in every cell of an organism. GPCRs are therefore involved in many diseases, since altered ligand or receptor levels and genetic or epigenetic modifications can lead to GPCR dysfunction and hence a pathophysiological phenotype.
About one third of currently marketed drugs target GPCRs. The human genome contains 720-800 predicted GPCRs, and about half of them respond to olfactory/sensory signals, whereas the others are known or predicted to be activated by endogenous ligands and many of these represent potential drug targets. Seventy seven per cent of these non-sensory GPCRs belong to the class A (rhodopsin-like) family, whereas 14 per cent represent class B (secretin-like) GPCRs, less than one per cent belong to the class C (metabotropic receptor-like) or the atypical frizzled-/smoothened receptor class, and the remaining 25 per cent are orphan receptors1,2.
All of these families share a common overall structure consisting of an extracellular aminoterminus, a seven helical transmembrane domain and an intracellular carboxy-terminus3 (Figure 1). The diverse class A family is generally activated by small molecules binding to a pocket in the transmembrane domain (TMD), while in class B the natural ligands are peptides which use the TMD pocket and the large N-terminal domain, and in family C the ligands bind to the N-terminal domain alone, causing conformational changes that trigger receptor signalling.
Drug binding sites can be located in the TMD, in the extracellular or intracellular loop regions, in larger extracellular domains, and may be orthosteric or allosteric with respect to the natural ligand binding sites4,5. Pharmaceutical lead identification and optimisation for GPCR drugs is completely dependent on cell-based assay systems (including cell membrane assays) because of limitations to obtain sufficient amounts of functional and stable protein for biochemical and biophysical assay methods. Such methods, however, are required to demonstrate on-target effects and to characterise the mechanism of action of compounds (or therapeutic antibodies) in an early lead discovery phase (Figure 2).
FIGURE 1 Structures of active and inactive conformations of the β2 adrenoceptor Structures of the β2 adrenoceptor (light grey) in an inactive conformation with the antagonist carazolol bound (left33), and in an active conformation in complex with Gs (dark grey) and the agonist BI-167107 bound (right41). In both cases, the ligand is represented by black spheres to show the location of the orthosteric binding site within the membrane-spanning region of the protein. T4 lysozyme (black) is inserted in the third intracellular loop between helices 5 and 6 in the inactive structure (bottom left), and fused in the extracellular N-terminus for the active structure (top right), to remove flexibility and to provide polar surfaces for crystallisation. The nanobody (black, bottom right) served to stabilise the open conformation of Gs and also provided crystallisation contacts
Significant progress has been made in recent years in the development of membrane mimetics for receptor stabilisation of bio – chemical and biophysical methods, and of methods to enable X-ray structural studies for GPCRs. This review gives an update and thoughts on how these new developments may revolutionise the classical cellular lead discovery of GPCRs in the future.
Classical cellular lead discovery for GPCRs
Current assay technologies used for screening of GPCRs are mainly based on cellular functional assays (Table 1). GPCRs signal via one or more types of heterotrimeric G proteins (αβγ. (Gs, Gi, Gq, and G12 families) and other cellular proteins6,7. Cellular assays applied in screening detect second messengers or changes in signalling proteins8, and the receptor function is measured in a cell-physiological context as compared to a biochemical assay. Such functional assays have proven to be successful in identifying GPCR-modulating compounds; however, one cannot be sure of on-target action of compounds. Moreover, due to the complex cellular environment it is not possible to gain a comprehensive mechanistic understanding of compound action.
Table 1: Most widely used assays in GPCR drug discovery*
*Cellular assays widely applied to GPCR screening include: measurement of intracellular Ca2+ using fluorescent dyes or photoproteins (and most Gi/Gs-GPCRs can be also linked e.g. via promiscuous Gα16), intracellular 3′,5′-cyclic adenosine monophosphate (cAMP) or inositol-monophosphate (IP1) using homogeneous immunocompetition assays, or indirect measurement of ligand-induced receptor internalization by e.g. detecting b-arrestin binding by enzyme fragment complementation of labeled b-arrestin to a labeled GPCR. Further secondary assay formats include measurement of phosphorylated/total ‘extracellular signal-regulated kinases’ (ERK) using specific bead-coupled antibodies, and various reporter gene assays [8].
| Readout | G Protein/Other | Type |
| Mobilization of intracellular Ca2+ | Gaq, Ga16, Gbg | Cellular |
| Accumulation of intracellular IP1 | Gaq, Ga16, Gbg | Cellular |
| Stimulation/inhibition of cAMP levels | Gas, Gai/o,, Gbg | Cellular |
| Receptor internalization (b-arrestin binding) | b arrestin | Cellular |
| Activation of ERK1/2 (phospho-ERK1/2) | Gbgi, b arrestin | Cellular |
| [35S]GTPγS SPA binding | Mainly Gai/o | Membrane-based |
| Radioligand SPA binding | Independent | Membrane-based |
The only biochemical assays typically applied in lead discovery are radioligand binding and [35S]guanosine-5’-O-(3’-thio)- triphosphate (GTPγS) binding assays using cell membranes8, i.e. not using purified protein. Bound radioactivity in these assays can be quantified using the bead-based homogeneous scintillation proximity assay (SPA) method9. Displacement binding assays utilise a radio – labelled orthosteric agonist (or antagonist), while GTPγS binding to Gα indirectly reflects G protein activation, and is generally limited to Gi/o-coupled receptors10. As these assays are based on cell membranes derived from transfected cell lines, it is again not possible to gain a comprehensive mechanistic understanding and to confirm on-target action with the exception of confirming orthosteric ligands in binding experiments. With the latter information, compounds are not filtered, but classified into ‘competitive and hence orthosteric’ ligands and into (the usually larger group of ) unknown compounds, which may bind to (a) an allosteric receptor pocket, (b) a receptor oligomer, or (c) do not bind to the GPCR target at all.
In initial high throughput screening (HTS) and subsequent orthogonal and secondary assays, engineered cells highly overexpressing the GPCR target are typically required to obtain robust signals (Figure 2). GPCR overexpression can lead to artificial signalling, and absolute compound activities that are related to the receptor expression level. In later lead discovery stages, lower compound numbers allow profiling in primary cells natively expressing the GPCR of interest. On-target action of compounds can only be confirmed if the natural (orthosteric) ligand can be radiolabelled, or an alternative synthetic ligand is available to measure binding affinities of orthosteric ligands. Exceptionally, an allosteric ligand might be available for labelling; however, further distinct allosteric binding sites cannot be excluded. The binding of allosteric ligands therefore cannot be measured with traditional ligand binding assays. More recently, a strong interest for allosteric ligands binding to a distinct receptor site has evolved, as these binding sites are generally less conserved among closely related receptors. Less lead optimisation efforts are consequently required to achieve selectivity of drug molecules.
Therefore, label-free methods based on isolated GPCR protein are required to confirm on-target action of orthosteric and allosteric ligands. Biochemical and biophysical methods using isolated GPCR protein are needed to study the mechanism of action of compounds to allow an early prioritisation of desired compound classes.
Recent technical developments using isolated GPCR proteins
The isolation of functional GPCRs from their native membrane in larger quantities remains a challenge due to factors such as protein aggregation, misfolding, glycosylation, or palmitoylation11, but useful amounts can be generally obtained. A major challenge has been the stability of solubilised GPCRs extracted from the cell membrane because detergents tend to destabilise the receptor. New membrane mimetics have evolved to solubilise and stabilise functional GPCR proteins in the absence of detergents (Table 2). These include recombinant amphipols, ‘high density lipoprotein’ (rHDL) particles, and ‘styrene maleic acid lipid particles’ (SMALP), which all mimic the native membrane environment by forming a lipid bilayer surrounding the GPCR12-14.
TABLE 2 Membrane mimetics for stabilisation of GPCR proteins
| Mimetic | Advantage | Disadvantage | Reference/s |
| rHDLs (Apo-A1) | Detergent-free, integration of G proteins shown | Requires purified GPCR (and Apo-A1) | [12,17-23] |
| SMALPs (Polymer) | Detergent-free, no GPCR purification required, reversible association | Not known | [12,16] |
| Amphipols (Polymer) | Detergent-free | Requires purified GPCR; limited utility of current polymers | [12,13,15] |
Amphipols are amphipathic polymers that hypercoil around the transmembrane regions of GPCRs. Although a number of isolated GPCRs have been shown to be functional using amphipols, the function of some of these proteins was impaired, and issues regarding polymer monodispersion and aggregation have been observed13,15. Current amphipols therefore appear to require further development for broader application.
SMALPs are another type of amphipathic polymers consisting of poly(styrene-co-maleic acid) (SMA), which reversibly surround a lipid bilayer of 10 nanometre diameter12,16. The SMA co-polymer can simply be added to cell membranes containing the GPCR target, and at a neutral or basic pH the particles self-assemble around GPCR protein and lipids from the cell membrane (or added exogenous lipids). At an acidic pH, the polymer is protonated, becomes insoluble and disassociates, and the protein membrane can thus be regenerated. As compared to amphipols and rHDLs, SMALPs do not require pre-purification of the GPCR protein, and hence no addition of deter – gent for solubilisation before removal for disc assembly. The technology might be a breakthrough as this is not only less laborious, but also most suitable for preserving folding and hence GPCR functionality.
Most of the functional data for GPCRs incorporated into membrane mimetics is reported for rHDLs (also called ‘nanodiscs’). rHDLs consist of wildtype or mutated apolipoprotein A1, an amphipathic protein, two of which irreversibly encapsulate a lipid bilayer of 10 nanometres14. Phospholipids are added to purified GPCR protein and apolipoprotein A1 solubilised in detergent, and membrane discs assemble upon removal of detergent. The rHDL membrane disc technology was already successfully applied to four mammalian class A GPCRs: rhodopsin, the β2 adrenoceptor, the μ opoid receptor and the CCR5 chemokine receptor17-23. When reconstituted in rHDLs they not only show the expected pharmacology in radioligand binding experiments, but also enable novel applications of biochemical and biophysical assays for isolated GPCRs (Table 3).
Table 3: Novel applications of biochemical and biophysical assays for GPCR proteins
*As examples, (high and low affinity GPCR agonists in the presence and absence of G protein, respectively, or receptor phosphorylation and b arrestin binding can be investigated. GTPgS binding can be measured from non-Gi/o-linked GPCRs such as (Gs-coupled) b2 adrenoceptors in rHDLs with additionally reconstituted Gprotein heterotrimer due to an improved signal-to-background ratio. Ligand affinities or potencies in GTPgS binding in the presence of different purified G protein subtypes (or b arrestin) can be examined to dissect pharmacology in a G protein-specific manner.
| Method | Material | Benefit | Reference/s |
| Radioligand Binding* | GPCR-rHDL (± Gx) | High/low ligand affinity (± G protein), G protein-specific. | [19,57] |
| [35S]GTPgS Binding* | GPCR-Gx-rHDL | G protein-specific, enabling for non-Gi/o-linked GPCRs. | [17,19-22] |
| SPR, Binding | Membrane/ Stabilized Protein, Immobilization | Label-free. Detection of allosteric ligands. Real-time kinetics. | [24-26] |
| BSI, Binding | Membrane/ Stabilized Protein | Label-free, no immobilization required. Detection of allosteric ligands. Real-time kinetics. | [27] |
Besides mechanistic studies in biochemical assays, isolated GPCR protein stabilised in membrane mimetics can be subjected to biophysical assays such as ‘surface plasmon resonance’ (SPR) or back-scattering interferometry (BSI). Such label-free binding assays have already been applied to cell membranes, such as immobilisation of a peptide or protein GPCR ligand for SPR has been reported24, but this has no advantage over a traditional radioligand binding assay due to the focus on orthosteric ligands. Alternatively, the Gα protein is immobilised, and binding of an activated GPCR measured25,26. Another biophysical binding technique is BSI, which detects changes in the refractive index of a solution. BSI requires less protein than SPR and no immobilisation, and the pharmacology of two GPCRs has been already proven27. Isolated and stabilised GPCR protein is needed to clearly confirm binding and therefore on-target action of orthosteric and allosteric compounds using biophysical assays such as SPR and BSI. Moreover, these assays provide real-time binding kinetics, i.e. ligand association/dissociation rates can be determined in addition to affinities measured at equilibrium.
Although both SMALPs and rHDLs are promising membrane mimetics for GPCRs, their broad applicability remains to be shown, and further development in this area may be needed. The evolving field of GPCR protein stabilisation highlights that tools and novel biochemical and biophysical assay applications are becoming available to close gaps in lead discovery with respect to mechanistic insights not retrievable from cell-based assays. Similarly, tools are becoming available to close the gap of GPCR structure determination, which is crucial for rational drug design besides purely empirical assay methods to facilitate and accelerate lead optimisation.
Structure-based drug discovery
A good understanding of the structure and function of a protein not only provides a basis for design and optimisation of novel chemical matter, it can enable virtual screening, fragmentbased drug design and also provide ideas for new approaches for target modulation. Up until 2007, the only experimental structural information available for a GPCR was from various states of rhodopsin3,28,29. Homology models built based on this information were essentially correct with respect to the positioning of helices in the TMD, but details in the ligand-binding pocket were difficult to predict largely due to variations in the extracellular loops, which we know to be very important for ligand binding30. Since then there have been structures determined for six different class A GPCRs (Table 4). This breakthrough is due to many recent developments in the production and handling of these highly flexible membrane proteins.
Table 4: Summary of available GPCR crystal structures*
* All GPCR structures available in the Protein Data Bank (http://www.rcsb.org/pdb) as of September 2011, except for Rhodopsin (~25 structures of various states), which is left out for space reasons. TSM = thermostabilising mutations, T4L = T4 lysozyme fusion, Cryst = crystallisation method, VD = vapour diffusion, LCP = lipidic cubic phases.
| GPCR | Modifi-cations | Cryst. | Ligand | Type | PDB | Ref. |
| Adenosine A2A Receptor | T4L | LCP | ZM241385 | antagonist | 3eml | [31] |
| T4L | LCP | UK-432097 | agonist | 3qak | [37] | |
| TSM | VD | adenosine | agonist | 2ydo | [58] | |
| TSM | VD | NECA | agonist | 2ydv | [58] | |
| TSM | VD | ZM241385 | inverse agonist | 3pwh | [59] | |
| TSM | VD | XAC | antagonist | 3rey | [59] | |
| TSM | VD | caffeine | antagonist | 3rfm | [59] | |
| b1 Adreno-ceptor | TSM | VD | cyanopindolol | antagonist | 2vt4 | [60] |
| TSM | VD | R-dobutamine | agonist | 2y00, 2y01 | [61] | |
| TSM | VD | R,R-carmoterol | agonist | 2y02 | [61] | |
| TSM | VD | R-isoprenaline | agonist | 2y03 | [61] | |
| TSM | VD | R-salbutamol | agonist | 2y04 | [61] | |
| TSM | VD | carazolol | antagonist | 2ycw | [62] | |
| TSM | VD | cyanopindolol | antagonist | 2ycx, 2ycy | [62] | |
| TSM | VD | iodocyano-pindolol | antagonist | 2ycz | [62] | |
| b2 Adreno-ceptor | Fab | Bicelles | carazalol | antagonist | 2r4r, 2r4s | [63] |
| T4L | LCP | carazalol | antagonist | 2rh1 | [33] | |
| T4L | LCP | timolol | antagonist | 3d4s | [34] | |
| Fab | Bicelles | – | – | 3kj6 | [50] | |
| T4L | LCP | ICI 118,551 | inverse agonist | 3ny8 | [42] | |
| T4L | LCP | JSZ | inverse agonist | 3ny9 | [42] | |
| T4L | LCP | alprenolol | antagonist | 3nya | [42] | |
| T4L, nanobody | LCP | BI-167107 | agonist | 3pog | [38] | |
| T4L | LCP | FAUC50 | irreversible agonist | 3pds | [39] | |
| T4L, nanobody | LCP | BI-167107, Gs-bound | agonist | 3sn6 | [41] | |
| CXCR4 | T4L | LCP | CVX15 | antagoinst | 3oe0 | [35] |
| T4L | LCP | IT1t | antagoinst | 3odu, 3oe6, 3oe8, 3oe9 | [35] | |
| Dopamine D3 Receptor | T4L | LCP | eticlopride | antagonist | 3pbl | [36] |
| Histamine H1 Receptor | T4L | LCP | doxepin | inverse agonist | 3rze | [40] |
Crystallisation requires a stable and homogeneous sample with respect to purity, but also with respect to conformational states. GPCRs are very unstable outside the membrane environment, are highly flexible as they need to adopt different conformational states to function and they are often post-translationally modified (e.g. glycosylation). To favour crystallisation, most GPCRs had to have flexible regions truncated (e.g. N-terminus, third intracellular loop and C-terminus) and for some, removal of glycosylation sites by site-directed mutagenesis was necessary. However, key to the recent successes was stabilisation of the GPCR in one particular conformational state by one or more of the following methods: thermostabilisation of the receptor, cocrystallisation with a stable binding partner, locking the conformational state with a high affinity ligand, or fusing a stable soluble protein (T4 lysozyme, T4L) into one of the loop regions. For example, in the case of the β1 adrenoceptor, thermostabilising mutations were determined by systematic mutagenesis and subsequent analysis of the binding of an antagonist, which led to the preferential adoption of the inactive conformation31. Thermostabilisation of GPCRs not only enables crystallisation, but also provides enough stability to allow coupling of the receptor to chips to determine kinetics of ligand binding by SPR32. In addition, since the receptor is less dependent on the affinity of the bound ligand for stabilisation, lower affinity compounds can be analysed by crystallography, which is an advantage for early stages of medicinal chemistry optimisation and allows the application of fragment-based drug discovery methods.
Many of the recent GPCR structures have been obtained by increasing the polar surface of the protein to provide greater potential for formation of crystal contacts31,33-42. This is done by inserting T4L into inner loop 3, which also reduces the conformational flexibility of this region. Another method of stabilising GPCR conformational states and providing extra polar surface areas for crystal contacts is to couple them with binding partners such as nanobodies, which are small (15 kDa), stable, single domain fragments of the single chain antibodies of Camelids43. This has been particularly successful for trapping the protein in an active state, where the nanobody acts as a surrogate for a G protein, and in the case of β2 adrenoceptor, increased the affinity of the GPCR for agonist binding by 100-fold38. However, it was a combination of many new developments that enabled the structure determination of an agonist-bound β2 adrenoceptor.in complex with a nucleotidefree Gs heterotrimer41. Other than the search for novel detergents for solubilisation and the use of a high affinity agonist to stabilise the complex, the use of single particle electron microscopy was critical to identify the position for fusion of T4L that gave a relatively fixed orientation of the insertion. It was also used to show that the the Gαs, helical domain was flexible in the absence of a nucleotide, but since the presence of guanosine-triphophate (GTP) or -diphosphate (GDP) would cause dissociation of the complex, a nanobody was generated to stabilise the conformation. Subsequent crystallisation in lipidic cubic phase (LCP) benefitted from the additional polar surfaces of both the T4L and the nanobody, leading to the first atomic resolution view of GPCR signalling41. It is import – ant to note that all of these manipulations of the GPCR to enable stabilisation and crystallisation have to be checked for functionality, in order to make sure that the resulting crystal structure is relevant for drug design44. Thermostabilisation of receptors enables them to be crystallised in the presence of detergents using vapour diffusion methods that are commonly used for soluble proteins. However, most of the non-thermostabilised receptors listed in Table 4 have been crystallised in lipidic cubic phases. The membrane-like environment formed by lipids such as the mono-oleins is more favourable for GPCRs than crystallisation detergents, which tend to be destabilising for membrane proteins. The lipid bilayer also favours order, which is good for high resolution diffraction, and it can filter out large impurities or aggregates, due to the need for long-range diffusion of the protein for crystal nucleation and growth. The latter can be optimised to increase the chances of crystallisation using fluorescence recovery after photo bleaching (FRAP), with a precrystallisation screening assay to optimise variables such as pH and salt prior to crystallisation trials. This rather expensive method has recently been automated to reduce the costs of time and protein45. Recent developments in detection of crystals growing in LCP, such as second order nonlinear imaging of chiral crystals (SONICC), will overcome the limitations of UV detection (poor signal to noise due to low intrinsic tryptophan content or heavy precipitation)46. Membrane protein crystals are usually very small (μm) and multiple crystals are required to get a full dataset due to radiation damage. This places stringent requirements on X-ray diffraction sources with respect to beam focus, goniometer accuracy, crystal centering, optimal data collection strategies and improved algorithms for processing data from multiple crystals. The development of free electron X-ray lasers (FEL), which offer 109 times more X-ray flux than the current synchrotron sources, will enable data collection from even smaller nanocrystals47. The technical advances in structural biology are necessary to increase coverage of the GPCR family, because although ligand docking with homology models is useful when sequence identity is high (35 per cent or more), docking predictions are poor for GPCRs with lower homology, when there are variations in kinks and helical structure within the transmembrane region and and for binding sites involving variable extracellular loops48. A comparison of TMD ligand binding sites between the available GPCR structures shows how dramatic the differences are in the shapes of the cavities and in the positions of the ligands within the sites49. This is largely due to extracellular loop 2, which is highly variable and very important for ligand binding. In the absence of crystal structures, site directed mutagenesis has been used successfully to gain information about which residues are important in order to improve the models, although care has to be taken not to interpret allosteric effects as direct contacts31. Nuclear magnetic resonance (NMR) can also be used to gain structural information about the extracellular loops. For example, selective 13C labelling of extracellular lysine residues in β2 adrenoceptor allowed the study of changes induced by antagonists and agonists compared to the unligated state and showed that there was a weakening of a salt bridge between extracellular loops 2 and 3 when agonist bound50. The reconstitution of the chemokine receptor CCR5 in rHDLs extends the lifetime of this protein enough to be able to map its interaction with the chemokine CCL3/MIP-1a23. The full structure determination of sensory rhodopsin II shows that GPCR structures can also be determined by NMR, if large amounts (milligrams) of isotopically labelled and stable protein can be produced51. A recent review of solution NMR applied to membrane proteins gives many examples of how this versatile technique is providing dynamic as well as structural and ligand interaction information, and how developments in the methodology, sample preparation and stabilisation will broaden its application in the future52.
The availability of a crystal structure for a target enables two other lead finding methods to be employed in parallel to HTS: virtual screening of compound libraries using computational high throughput docking, and fragment-based screening (FBS). Virtual screening can not only discover compounds missed in HTS, but can be employed to external libraries to purchase additional compounds for experimental testing53. As one among many examples, new chemical classes of β2 adrenoceptor antagonists were found by this method54. FBS by biochemical or biophysical methods (e.g. NMR, SPR) aims to identify small fragments (<250Da) with a high ligand efficiency that will be ideal starting points for chemical optimisation because of their small size55. This method has been shown to be successful in identifying hits binding to allosteric sites of soluble proteins56. The thermo – stabilisation of receptors by site directed mutagenesis could enable FBS for GPCRs by making them suitable for FBS by SPR and by providing a means of getting crystal structures with weaker ligands, which is essential to provide directions for medicinal chemistry optimisation32.
Future perspectives
The evolution of novel tools for stabilisation of isolated GPCR proteins for biochemical, biophysical, and structural studies will boost the lead discovery success for this therapeutically highly important target class.
Currently, for compound classes active in cellular screening assays, it is often not possible to predict why some classes are active in primary cell and in vivo animal models while others are not. Mechanistic insights using isolated GPCR (and G) proteins are therefore required to elucidate the receptor pharmacology in a G protein-dependent (and independent) manner. Better characterisation of the binding modes will facilitate discovery of novel allosteric ligands to explore chemical space beyond classical orthosteric drugs. Large quantities of stabilised, isolated GPCRs are currently out of scope for high throughput screening, so biochemical assays will not replace cellular assays as for soluble proteins. Nevertheless, quantities are sufficient to mechanistically characterise a smaller set of compounds found in cellular assays to facilitate prioritisation of chemistry efforts to most promising scaffolds, and consequently to lower the attrition rate of classes in later lead optimisation.
Despite the fact that the efforts required to obtain novel GPCR structures will remain quite high, there is no doubt that structural coverage within class A and beyond will increase. Novel structural information, especially for allosteric ligands and in class B and C GPCRs will provide a breakthrough for the discovery of drugs for important clinical GPCR targets for which classical methods have not produced suitable compounds so far. A better structural understanding of how GPCRs signal through G and other proteins could also provide novel approaches for lead discovery, for example by finding compounds that specifically block the interaction with downstream partners. The upcoming decade will bear witness to exciting developments in lead discovery for GPCRs, which we anticipate will accelerate novel GPCR-targeting drugs entering the clinic.
References
1. Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, Brown A, Rodriguez SS, Weller JR, Wright AC, Bergmann JE, Gaitanaris GA. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903-4908
2. Fredholm BB, Hoekfelt T, and Milligan G. G-proteincoupled receptors: an update. Acta Physiol. 2007, 190, 3–7
3. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, and Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 2000, 289, 739–745
4. Kristiansen K. Molecular mechanisms of ligand binding, signalling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Therap. 2004, 103, 21– 80
5. Salchow K, Bond ME, Evans SC, Press NJ, Charlton SJ, Hunt PA, Bradley ME. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharmacol. 2010, 159, 1429-1439
6. Marinissen MJ, and Gutkind JS. G-protein-coupled receptors and signalling networks: emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368-376
7. Offermanns S. G proteins as transducers in transmembrane signalling. Prog. Biophys. Mol. Biol. 2003, 83, 101-130
8. Siehler S. Cell-based assays in GPCR drug discovery. Biotechnology J. 2008, 4, 471-483
9. Alouani S. Scintillation proximity binding assay. Methods Mol. Biol. 2000, 138, 135-141
10. Milligan G. Principles: extending the utility of [35S]GTPγS binding assays. Trends Pharmacol. Sci. 2003, 24, 87–90
11. Chiu ML, Tsang C, Grihalde N, MacWilliams MP. Overexpression, solubilization, and purification of G proteincoupled receptors for structural biology. Comb. Chem. High Throughput Screening 2008, 11, 439-462
12. Jamshad M, Lin YP, Knowles TJ, Parslow RA, Harris C, Wheatley M, Poyner DR, Bill RM, Thomas OR, Overduin M, Dafforn TR. Surfactant-free purification of membrane proteins with intact native membrane environment. Biochem Soc Trans. 2011, 39, 813-818
13. Dahmane T, Damian M, Mary S, Popot JL, Banères JL. Amphipol-assisted in vitro folding of G protein-coupled receptors. Biochemistry 2009, 48, 6516-6521
14. Shih AY, Denisov IG, Phillips JC, Sligar SG, Schulten K. Molecular dynamics simulations of discoidal bilayers assembled from truncated human lipoproteins. Biophys. J. 2005, 88, 548-556
15. Popot JL. Amphipols, nanodiscs, and fluorinated surfactants: three nonconventional approaches to studying membrane proteins in aqueous solutions. Annu. Rev. Biochem. 2010, 79, 737–775
16. Knowles T J, Finka R, Smith C, Lin YP, Dafforn T, Overduin M. Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J. Am. Chem. Soc. 2009, 131, 7484–7485
17. Leitz AJ, Bayburt TH, Barnakov AN, Springer BA, Sligar SG. Functional reconstitution of β2-adrenergic receptors utilizing self-assembling nanodisc technology. Biotechniques 2006, 40, 601-602
18. Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J. Biol. Chem,. 2007, 282, 14875-14881
19. Whorton MR, Bokoch MP, Rasmussen SGF, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G proteincoupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U S A 2007, 104, 7682-7687
20. Whorton MR, Jastrzebska B, Park PSH, Fotiadis D, Engel A, Palczewski K, Sunahara RK. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 2008, 283, 4387-4394
21. Kuszak AJ, Pitchiaya S, Anand JP, Mosberg HI, Walter NG, Sunahara RK. Purification and functional reconstitution of monomeric μ-opioid receptors: allosteric modulation of agonist binding by Gi2. J. Biol. Chem. 2009, 284, 26732-26741
22. Yao XJ, Vélez Ruiz G, Whorton MR, Rasmussen SG, DeVree BT, Deupi X, Sunahara RK, Kobilka B. The effect of ligand efficacy on the formation and stability of a GPCRG protein complex. Proc. Natl. Acad. Sci. U S A 2009, 106, 9501-9506
23. Yoshiura C, Kofuku Y, Ueda T, Mase Y, Yokogawa M, Osawa M, Terashima Y, Matsushima K, Shimada I. NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers. J. Am. Chem. Soc. 2010, 132, 6768-6777
24. Harding PJ, Attrill H, Ross S, Koeppe JR, Kapanidis AN, Watts A. Neurotensin receptor type 1: Escherichia coli expression, purification, characterization and biophysical study. Biochem Soc Trans. 2007, 35, 760-763
25. Alves ID, Park CK, Hruby VJ. Plasmon resonance methods in GPCR signalling and other membrane events. Curr. Protein Pept. Sci. 2005, 6, 293-312
26. Komolov KE, Senin II, Philippov PP, Koch KW. Surface plasmon resonance study of G protein/receptor coupling in a lipid bilayer-free system. Anal Chem. 2006, 78, 1228-1234
27. Baksh MM, Kussrow AK, Mileni M, Finn MG, Bornhop DJ. Label-free quantification of membrane-ligand interactions using backscattering interferometry. Nat. Biotechnol. 2011, 29, 357-360
28. Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J. Mol. Biol. 2007, 372, 1179-1188
29. Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 16123-16128
30. Peeters MC, van Westen GJ, Li Q, IJzerman AP. Importance of the extracellular loops in G proteincoupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35-42
31. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211-1217
32. Robertson N, Jazayeri A, Errey J, Baig A, Hurrell E, Zhukov A, Langmead CJ, Weir M, Marshall FH. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology 2011, 60, 36-44
33. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G proteincoupled receptor. Science 2007, 318, 1258-1265
34. Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897-905
35. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066-1071
36. Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091-1095
37. Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322-327
38. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 2011, 469, 175-180
39. Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK. Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 2011, 469, 236-240
40. Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65-70
41. Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β(2) adrenergic receptor-Gs protein complex. Nature 2011, doi: 10.1038/ nature10361
42. Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved Binding Mode of Human β2 Adrenergic Receptor Inverse Agonists and Antagonist Revealed by X-ray Crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445
43. Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 2011, 21, 567-572
44. Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 2009, 19, 386-395
45. Xu F, Liu W, Hanson MA, Stevens RC, Cherezov V. Development of an Automated High Throughput LCPFRAP Assay to Guide Membrane Protein Crystallization in Lipid Mesophases. Cryst. Growth Des. 2011, 11, 1193-1201
46. Kissick DJ, Gualtieri EJ, Simpson GJ, Cherezov V. Nonlinear optical imaging of integral membrane protein crystals in lipidic mesophases. Anal. Chem. 2010, 82, 491-497
47. Fromme P, Spence JCh. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol. 2011, 21, 509-516
48. Kufareva I, Rueda M, Katritch V; GPCR Dock 2010 participants, Stevens RC, Abagyan R. Status of GPCR Modeling and Docking as Reflected by Communitywide GPCR Dock 2010 Assessment. Structure 2011, 19, 1108-1126
49. Congreve M, Langmead CJ, Mason JS, Marshall FH. Progress in structure based drug design for G proteincoupled receptors. J. Med. Chem. 2011, 54, 4283-4311
50. Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, Puglisi JD, Weis WI, Pardo L, Prosser RS, Mueller L, Kobilka BK. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010, 463, 108-12
51. Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach S. Structure determination of the sevenhelix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nature Struct. Mol. Biol. 2010, 17, 768-774
52. Nietlispach D, Gautier A. Solution NMR studies of polytopic α-helical membrane proteins. Curr. Opin. Struct. Biol. 2011, 21, 497-508
53. Vilar S, Ferino G, Phatak SS, Berk B, Cavasotto CN, Costanzi S. Docking-based virtual screening for ligands of G protein-coupled receptors: Not only crystal structures but also in silico models. J. Mol. Graph. Model. 2011, 29, 614-623
54. Sabio M, Jones K, Topiol S. Use of the beta2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Biorg. Med. Chem. Lett. 2008, 18, 5391-5395
55. Schultes S, de Graaf C, Haaksma EEJ, de Esch IJP, Leurs R, Kraemer O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today: Technologies 2010, 7, E157-E162
56. Jahnke W, Rondeau JM, Cotesta S, Marzinzik A, Pellé X, Geiser M, Strauss A, Götte M, Bitsch F, Hemmig R, Henry C, Lehmann S, Glickman JF, Roddy TP, Stout SJ, Green JR. Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat. Chem. Biol. 2010, 6, 660-666
57. Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin- 1 binding. J. Biol. Chem. 2011, 286, 1420-1428
58. Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521-525
59. Doré AS, 3, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, Hurrell E, Bennett K, Congreve M, Magnani F, Tate CG, Weir M, Marshall FH. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283-1293
60. Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-proteincoupled receptor. Nature 2008, 454, 486-491
61. Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature 2011, 469, 241-244
62. Moukhametzianov R, Warne T, Edwards PC, Serrano- Vega MJ, Leslie AG, Tate CG, Schertler GF. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 8228-32
63. Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383-387
About the Authors
Dr. Sandra Siehler studied Biology and received her PhD at ETH Zurich in 1999. Following postdoctoral research at the University of Pennsylvania, Philadelphia, she started as a Laboratory Head at Novartis in Basel in 2002. She focuses on assays and innovative research for GPCRs, and is experienced in leading drug discovery programs.
Dr. Sandra W. Cowan-Jacob completed her PhD in structural biology at the St. Vincents Institute of Medical Research and the University of Melbourne in Australia in 1988. She then did postdoctoral studies in Uppsala University, Sweden, and in the University of Basel, Switzerland, where she gained experience determining crystal structures of membrane proteins. Since 1993, she has been working in Ciba-Geigy and now Novartis, where she is now leading a group that applies X-ray, NMR and biophysics in lead discovery and optimisation projects.