

Treatment for rare Duchenne muscular dystrophy mutation approved by FDA
Posted: 26 February 2021 | Hannah Balfour (European Pharmaceutical Review) | 3 comments
The FDA conditionally approved Sarepta’s Amondys 45 (casimersen) after interim Phase III results indicated the treatment is reasonably likely to be of clinical benefit.
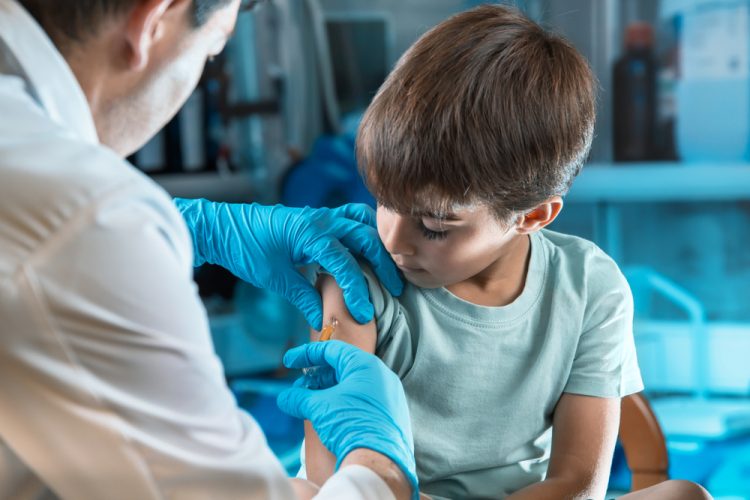
The US Food and Drug Administration has conditionally approved Amondys 45 (casimersen) injection for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping. This is the first FDA-approved targeted treatment for the eight percent of DMD patients with this type of mutation.
DMD is the most common type of muscular dystrophy, a rare genetic disorder characterised by progressive muscle deterioration and weakness. It is caused by mutations in the DMD gene which results in an absence of dystrophin, a protein found in muscle fibres. The first symptoms are usually seen between three and five years of age and worsen over time. The disease occurs in approximately one of every 3,600 male infants worldwide and can, in very rare cases, affect females.
Amondys 45 uses Sarepta Therapeutics’s proprietary phosphorodiamidate morpholino oligomer (PMO) chemistry and exon-skipping technology to direct the muscle cells to skip exon 45 when processing the messenger RNA molecule that cells use to make dystrophin protein. When exon 45 is skipped, the remaining exons assemble into a shorter, but still functional dystrophin protein. It is thought, as a result, that the treatment could potentially slow the decline in muscle strength.
The FDA approval was based on interim data from the Phase III ESSENCE (NCT02500381) trial assessing whether Amondys 45 (for exon 45 skipping) and Vyondys 53 (for exon 53 skipping) can significantly improve muscle strength. The trial enrolled 222 boys, ages seven to 13 with DMD amenable to either exon 45 or exon 53 skipping, to receive either active treatment or a placebo as a once-weekly infusion into the bloodstream for up to 96 weeks. This randomised phase will be followed by an open-label extension phase where all patients receive active treatment for up to 48 weeks.
The secondary endpoints of the trial include changes in dystrophin protein production and respiratory strength. The trial is expected to finish in May 2023.
The interim analysis includes 43 patients who were randomised 2:1 to receive either intravenous Amondys 45 (30 mg/kg) or placebo. After 48 weeks the analysis shows mean dystrophin protein levels in the Amondys 45 treatment group increased by a statistically significant 1.736 percent compared with 0.925 percent in the placebo group. Additionally, a sample of 22 Amondys 45 recipients showed an increase in the levels of mRNA in which exon 45 was skipped compared with their baseline levels. This represented a 100 percent response rate. Moreover, there was a statistically significant positive correlation between exon 45 skipping and dystrophin protein production.
The FDA concluded that the data submitted by the Serepta demonstrated an increase in dystrophin production that is reasonably likely to be of clinical benefit in patients with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 45 skipping. However, the exact clinical benefit, including whether the drug can improve motor function, is yet to be established. In making its decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease and the lack of available therapy.
The most common side effects observed in over 20 percent of DMD patients treated with Amondys 45 were upper respiratory tract infections, cough, fever, headache, joint pain and throat pain.
Although kidney toxicity was not observed in the Amondys 45 clinical studies, kidney toxicity was observed in the nonclinical studies and could include potentially fatal glomerulonephritis, which has been observed after administration of some antisense oligonucleotides. Therefore, kidney function should be monitored in patients taking Amondys 45.
“Developing drugs designed for patients with specific mutations is a critical part of personalized medicine,” said Dr Eric Bastings, deputy director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research. “Today’s approval of Amondys 45 provides a targeted treatment option for Duchenne muscular dystrophy patients with this confirmed mutation.”
A Phase III, open-label, extension study (NCT03532542) is enrolling 260 DMD male participants, ages seven to 23, by invitation. Its aim is to evaluate the long-term effects of Amondys 45 and Vyondys 53. Researchers expect to complete it in August 2026.
Related topics
Clinical Trials, Drug Development, Drug Safety, Proteins, Proteomics, Regulation & Legislation, Therapeutics



My younger brother is suffering from this issue of DMD. Kindly let me know if there is any treatment for this disease
I also want to know… Pls lemme know
My son is 12 years old suffering from DMD last 11 months he is not able to stand and walk we want to know the treatment in future
Please let us know
Thanks,
V. K. Bidri