

FDA permits marketing of AI-based device to detect diabetic retinopathy
Posted: 16 April 2018 | Dr Zara Kassam (European Pharmaceutical Review) | 1 comment
The FDA has permitted marketing of the first medical device to use artificial intelligence to detect greater than a mild level of the eye disease diabetic retinopathy…
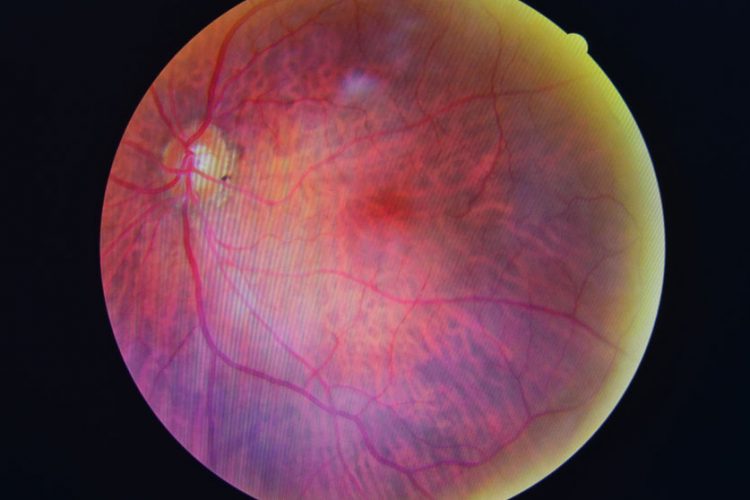
The US Food and Drug Administration has permitted marketing of the first medical device to use artificial intelligence to detect greater than a mild level of the eye disease diabetic retinopathy in adults who have diabetes.
Diabetic retinopathy occurs when high levels of blood sugar lead to damage in the blood vessels of the retina, the light-sensitive tissue in the back of the eye. Diabetic retinopathy is the most common cause of vision loss among the more than 30 million Americans living with diabetes and the leading cause of vision impairment and blindness among working-age adults.
“Early detection of retinopathy is an important part of managing care for the millions of people with diabetes, yet many patients with diabetes are not adequately screened for diabetic retinopathy since about 50 percent of them do not see their eye doctor on a yearly basis,” said Dr Malvina Eydelman, director of the Division of Ophthalmic, and Ear, Nose and Throat Devices at the FDA’s Center for Devices and Radiological Health. “Today’s decision permits the marketing of a novel artificial intelligence technology that can be used in a primary care doctor’s office. The FDA will continue to facilitate the availability of safe and effective digital health devices that may improve patient access to needed health care.”
The device, called IDx-DR, is a software program that uses an artificial intelligence algorithm to analyse images of the eye taken with a retinal camera called the Topcon NW400. A doctor uploads the digital images of the patient’s retinas to a cloud server on which IDx-DR software is installed. If the images are of sufficient quality, the software provides the doctor with one of two results: (01) “more than mild diabetic retinopathy detected: refer to an eye care professional” or (2) “negative for more than mild diabetic retinopathy; rescreen in 12 months.” If a positive result is detected, patients should see an eye care provider for further diagnostic evaluation and possible treatment as soon as possible.
IDx-DR is the first device authorised for marketing that provides a screening decision without the need for a clinician to also interpret the image or results, which makes it usable by health care providers who may not normally be involved in eye care.
The FDA evaluated data from a clinical study of retinal images obtained from 900 patients with diabetes at 10 primary care sites. The study was designed to evaluate how often IDx-DR could accurately detect patients with more than mild diabetic retinopathy. In the study, IDx-DR was able to correctly identify the presence of more than mild diabetic retinopathy 87.4 percent of the time and was able to correctly identify those patients who did not have more than mild diabetic retinopathy 89.5 percent of the time.
Patients who have a history of laser treatment, surgery or injections in the eye or who have any of the following conditions should not be screened for diabetic retinopathy with IDx-DR: persistent vision loss, blurred vision, floaters, previously diagnosed macular edema, severe non-proliferative retinopathy, proliferative retinopathy, radiation retinopathy or retinal vein occlusion. IDx-DR should not be used in patients with diabetes who are pregnant; diabetic retinopathy can progress very rapidly during pregnancy and IDx-DR is not intended to evaluate rapidly progressive diabetic retinopathy. IDx-DR is only designed to detect diabetic retinopathy, including macular oedema; it should not be used to detect any other disease or condition. Patients will still need to get a complete eye examination at the age of 40 and at the age of 60 and also if they have any vision symptoms (for example, persistent vision loss, blurred vision or floaters).
IDx-DR was reviewed under the FDA’s De Novo premarket review pathway, a regulatory pathway for some low- to moderate-risk devices that are novel and for which there is no prior legally marketed device. IDx-DR was granted Breakthrough Device designation, meaning the FDA provided intensive interaction and guidance to the company on efficient device development, to expedite evidence generation and the agency’s review of the device. To qualify for such designation, a device must provide for more effective treatment or diagnosis of a life-threatening or irreversibly debilitating disease or condition, and meet one of the following criteria: the device must represent a breakthrough technology; there must be no approved or cleared alternatives; the device must offer significant advantages over existing approved or cleared alternatives; or the availability of the device is in the best interest of patients.
The FDA is permitting marketing of IDx-DR to IDx LLC.



Much information provided about retinopathy, this is good to know.
Thanks for this post.