

Thermodynamic probes of cold instability: application to therapeutic proteins
Posted: 18 November 2019 | Bernardo Perez-Ramirez (Sanofi), Robert Simler (Amgen) | No comments yet
Developing a stable therapeutic protein formulation requires an intimate knowledge of the protein and its physical and chemical properties. In this article, Bernardo Perez-Ramirez and Robert Simler discuss the thermodynamic consequences that low temperature can have on the aggregation tendencies of a protein.

Abstract
Proteins are dynamic entities, constantly adopting different partially-folded states as a function of temperature and other solution variables. These variables dictate the standard free energy between the native, unfolded and partially folded ‘aggregation’ – prone states leading to oligomerisation. As a result, not all oligomerisation events in proteins are similar. Altering these variables, such as temperature, pH, salt and ligands, could induce a protein to ‘aggregate’ as a consequence of ‘unnatural folding’ to balance the thermodynamically unfavourable interactions between solvent and exposed hydrophobic residues in proteins. In the same way, these variables may induce a protein to self-associate, in mostly the native state, to counteract the unfavourable interactions with the solvent. Thus, cold instability without inducing cold denaturation could destabilise the native state of a protein, making it more prone to aggregation events.
Introduction
By calculating the free energy of folding at different denaturant concentrations, one can extrapolate back to determine the free energy of folding in the absence of a denaturant, ΔG°water“
The response of a protein’s properties to changes in temperature has long been one of the most useful probes when studying protein stability. Most of these studies have investigated the behaviour of a protein as temperature is increased, wherein the entropic contribution of the protein’s free energy favouring the more disordered unfolded form overtakes the enthalpic contribution that favours the compact native state with solvent excluded from the hydrophobic core. Such experiments, as monitored by differential scanning calorimetry or by a spectroscopic probe, provide direct measurements of the thermodynamic parameters ΔG, ΔH, ΔCp and ΔS. Additionally, in the pharmaceutical setting, accelerated stability studies, conducted at elevated but non-denaturing temperatures, are crucial in predicting likely degradation products during long-term storage.1 In these studies, potential biotherapeutics are incubated at temperatures higher than those intended for storage to destabilise the molecule and accelerate the formation of breakdown products, such as aggregates, that are likely to be observed on longer timescales at lower temperatures.
It has long been known that low temperature is just as effective at destabilising and denaturing a protein as high temperature.2-8 The theoretical explanation for this is rooted in a modified version of the Gibbs-Helmholtz equation to determine the Gibbs free energy at a particular temperature, ΔG(T):
![]()
where Tr is a reference temperature. The positive nature of the change in heat capacity, ΔCp, leads to a parabolic relationship between ΔG and temperature where there is a clear temperature of maximum stability and a high and low temperature boundary where the free energy of the protein favours the unfolded state.9 The shape and curvature of this free energy profile is specific to each individual protein and based on its specific thermodynamic parameters. The long-held thermodynamic explanation for the loss of stability, and eventually structure, at low temperatures is that the hydrophobic interactions within the protein are lessened due to favourable interactions between water and nonpolar groups at lower temperatures. This allows the entropy (ΔS) of the unfolded molecule to become more dominant than the enthalpy (ΔH) associated with the folded structure.
While the phenomenon of cold denaturation and dependence of folding free energy on temperature has been known for over 40 years,2 recent studies have shed new light on its importance. The cold‑denaturation mechanism had always been assumed to be an ‘all or nothing’ event, where the folded protein would completely unfold or not.10 However, cold denaturation is not entirely co‑operative and can also be used to study the partially-folded substructures of proteins.11,12 Like proteins at room temperature, these partially‑folded states are transiently sampled in the native ensemble.
Stability studies at higher temperatures may not always predict protein behaviour at refrigerated conditions
Accelerated stability studies at higher temperatures are a key component of formulation and drug product development of a potential biotherapeutic, particularly in the early pre-formulation stages.1,13 While these studies are usually supplemented with a real-time stability study at the lower temperature intended for storage, subsequent experiments are often designed primarily using these accelerated stability studies as a guide under the assumption that the same degradation products will be seen, albeit on longer timescales, at the lower storage temperature. This assumption is often, but not universally, true. This loss in stability as a result of low temperature can facilitate the acceleration of aggregate formation under these conditions.
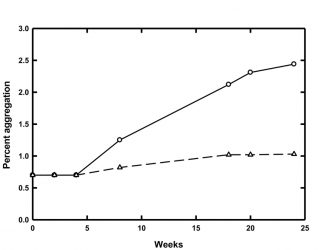
Figure 1: Percent of aggregated material for MG at 4°C (circles, solid line) and 8°C (triangles, dashed line) over 24 weeks of storage
One example of potential protein stability issues at low temperatures comes from our laboratory. Accelerated stability studies carried out at 25°C predicted that a model glycoprotein (MG) would be stable and resistant to aggregation in a particular buffer when stored at 2-10°C. However, real-time stability studies at 2-10°C showed that MG did in fact aggregate at these lower temperatures. Further studies revealed that the rate of aggregation was temperature dependent and that the lower the storage temperature, the faster the aggregation (Figure 1). MG tended to aggregate almost three times as quickly when stored at 4°C as opposed to 8°C.
In an effort to ascertain an underlying cause for this behaviour and consequently elucidate potential methods to prevent this aggregation, chemical denaturation experiments coupled with fluorescence spectroscopy were employed to determine the thermodynamic stability of MG as a function of temperature. The fluorescence spectra of MG in the absence and presence of 4 M guanidine hydrochloride are shown in Figure 2.
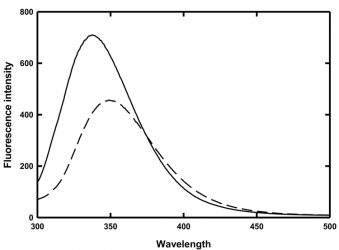
Figure 2: Fluorescence spectra of MG in the absence (solid line) and presence (dashed line) of 4 M guanidine hydrochloride
The folded and denatured forms of MG have wavelengths of maximum emission at 337nm and 350nm, respectively. Thus, monitoring the change in the ratio of the peak heights at 330nm and 350nm provides a means to observe the protein unfolding. Fluorescence intensities at a chosen wavelength have also historically been used to monitor protein folding, but those intensities are temperature dependent and thus in a study such as this, the use of fluorescence intensity would only introduce another unknown variable.
Chemical denaturation and elucidation of thermodynamic parameters
Chemical denaturation experiments have long been used to determine thermodynamic properties of proteins and were critical in elucidating the temperature dependence of the free energy of folding for proteins.9,14,15 In these experiments, the unfolding of a protein is monitored as increasing amounts of a chemical denaturant, in this case guanidine hydrochloride, are added. The linear extrapolation method16 can then be used to obtain a measurement of the Gibbs free energy of folding, ΔG°, for the protein. This method relies on the experimental relationship that the free energy of folding at a particular denaturant concentration varies linearly with the concentration of the denaturant. This leads to the equation:
![]()
By calculating the free energy of folding at different denaturant concentrations, one can extrapolate back to determine the free energy of folding in the absence of a denaturant, ΔG°water. The slope of the dependence of the free energy on concentration of denaturant is known as the m value and reflects the amount of buried surface area that becomes solventexposed upon unfolding.17 This value is useful when comparing the unfolding of a protein under different formulation conditions as, if one assumes that the unfolded state of the protein is the same under any condition, then a change in the m value would reflect a change in the structure of the protein in its native state as the result of a change in formulation.
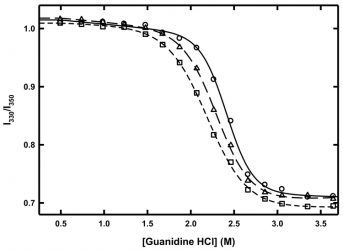
Figure 3: Chemical denaturation of MG at 10°C (circles, solid line), 6°C triangles, long dash line) and 2°C (squared, short-dash line). Lines represent fits to a two-state folding model.
Such chemical denaturation experiments can be conducted at different temperatures in order to determine the temperature dependence of the Gibbs free energy of unfolding. In the case of MG, chemical denaturation experiments were conducted at 10°C, 6°C and 2°C (Figure 3). As the temperature decreased, the amount of guanidine hydrochloride needed to completely unfold MG also decreased, indicating that the protein was becoming less thermodynamically stable at these low temperatures. When the curves were fitted to a two‑state unfolding model,18 the value of ΔG decreased by approximately 25 percent from 10°C to 2°C, while the m-values of the denaturation at 10°C, 6°C and 2°C were all within error of each other. The m-value result indicates that there was no change in the amount of buried surface area exposed upon chemical denaturation as a result of temperature and thus no cold denaturation is occurring. However, there is a clear loss in thermodynamic stability as a result of lower temperature.
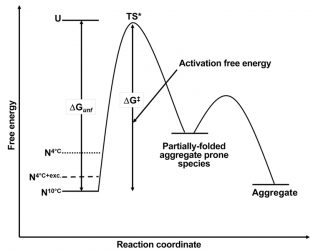
Figure 4: Schematic free energy diagram for the aggregation of MG at 10°C (N10°C, solid line), 4°C (N4°C, dotted line) and 4°C in the presence of a stabilising excipient (N4°C+ exc.). ΔGunf and ΔG‡ are the free energy of unfolding and the activation free energy respectively. TS* is the transition state between the native form of MG and the partially-folded aggregate-prone species.
Given the supposition that aggregation rates are related to the free energy difference of the native form of a protein with respect to its completely denatured form, these findings explain the potential root cause of the temperature dependence on aggregation rates within the 2-10°C range for MG. Building on the free energy diagram based on the Lumry-Eyring framework model for aggregation19 (Figure 4), the difference in free energy between the native state and the unfolded state for MG has been decreased as a result of lowering the temperature from 10°C to 2°C. As a result, the free energy difference between the native state at these lower temperatures and the partially-folded aggregationprone form of the protein has potentially also been decreased. While this aggregate-prone state, mostly in its native form, is still only sampled transiently, it is now being sampled more often at the lower temperatures and is thus available in greater frequency to facilitate an aggregation event. Hence, aggregation proceeds faster as the temperature is lowered.
A decrease in temperature can lead to enough of a loss of thermodynamic stability (cold instability) to facilitate the aggregation pathway”
Such knowledge is useful as it can also provide a methodology to predict ways to mitigate this acceleration in aggregation. In the case of MG, the addition of sucrose provides a 15 percent increase in the value of ΔG as determined by chemical denaturation experiments (data not shown). When this excipient is added during real-time stability studies at 4°C, the rate of aggregation slows significantly compared to that in the absence of the excipient (data not shown). With respect to the free energy diagram for MG (Figure 4), the addition of sucrose stabilises the native state of MG compared to the unfolded state as well as the partially-folded aggregate state, thus reducing the frequency that the MG molecule samples the aggregate-prone state and therefore decelerating the rate of aggregation.
The example shown here with MG illustrates the thermodynamic consequences that low temperature can have on the aggregation tendencies of a protein. While the assumption that most proteins will likely aggregate slower at 2-10°C than at 25°C is likely a good one, it is important to keep in mind that thermodynamic stability is temperature dependent. Thus, a decrease in temperature can lead to enough of a loss of thermodynamic stability (cold instability) to facilitate the aggregation pathway.
About the authors
 Bernardo Perez-Ramirez is currently Senior Scientific Director and Global Head of Marketed Products Life Cycle Management and Drug Product Due Diligence within Biologics Drug Product Development & Manufacturing at Sanofi in Framingham, Massachusetts. He is a protein biochemist and biophysicist by training and has worked for more than 25 years in pre-formulation research, formulation development, drug device integration, drug delivery strategies and technology evaluation. He began his career at Genetics Institute (now Pfizer), joining Genzyme Sanofi in 2004. He serves as Adjunct Professor at Tufts University and Honorary Professor of Formulation and Drug Product Development at the University of Nottingham, UK.
Bernardo Perez-Ramirez is currently Senior Scientific Director and Global Head of Marketed Products Life Cycle Management and Drug Product Due Diligence within Biologics Drug Product Development & Manufacturing at Sanofi in Framingham, Massachusetts. He is a protein biochemist and biophysicist by training and has worked for more than 25 years in pre-formulation research, formulation development, drug device integration, drug delivery strategies and technology evaluation. He began his career at Genetics Institute (now Pfizer), joining Genzyme Sanofi in 2004. He serves as Adjunct Professor at Tufts University and Honorary Professor of Formulation and Drug Product Development at the University of Nottingham, UK.
 B Robert Simler is currently a Director of Process Development in the Drug Product Technologies department at Amgen in Cambridge, Massachusetts where he oversees global commercial drug product development and lifecycle management activities. Prior to joining Amgen, Dr Simler held roles at Genzyme/Sanofi and Biogen where he was responsible for drug product formulation and process development spanning multiple modalities and all clinical and commercial phases.
B Robert Simler is currently a Director of Process Development in the Drug Product Technologies department at Amgen in Cambridge, Massachusetts where he oversees global commercial drug product development and lifecycle management activities. Prior to joining Amgen, Dr Simler held roles at Genzyme/Sanofi and Biogen where he was responsible for drug product formulation and process development spanning multiple modalities and all clinical and commercial phases.
References
- Guziewicz NG, Trierweiler G, Johnson K, Simler R, Perez-Ramirez B. Probing the solution behavior of therapeutic proteins by differential scanning calorimetry: Applications to pre-formulation and formulation development. In: Current Trends in Microcalorimetry. 2007. Boston MA, Microcal, LLC.
- Hopkins FG. (1930). Denaturation of proteins by urea and related substances. Nature, 126, 383.
- Chi Y, Kumar TK, Wang HM, Ho MC, Chiu IM, Yu C. (2001). Thermodynamic characterization of the human acid fibroblast growth factor: evidence for cold denaturation. Biochemistry, 40 (25) 7746-7753.
- Franks F, Hatley RH, Friedman HL. (1988). The thermodynamics of protein stability. Cold destabilization as a general phenomenon. Biophys Chem, 31, (3) 307-315.
- Hallerbach B, Hinz HJ. (1999) Protein heat capacity: Inconsistencies in the current view of cold denaturation. Biophys Chem, 76 (3) 219-227.
- Privalov PL, Griko, Yu, Venyaminov S, Kutyshenko VP. (1986). Cold denaturation of myoglobin. Mol. Biol. 190, (3) 487-498.
- Richardson JM, Leamaire SD, Jacquet JP, Makhatadze GI. (2000). Difference in the mechanisms of the cold and heat induced unfolding of thioredoxin h from Chlamydomonas reinhardii: spectroscopic and calorimetric studies. Biochemistry, 39, (36) 11154-11162.
- Lazar KL, Patapoff TW, Sharma V. (2010). Cold denaturation of monoclonal antibodies mAbs 2 (1), 45-52.
- Kumar S, Tsai CJ, Nussinov R. (2003). Temperature range of thermodynamic stability for the native state of reversible two-state proteins. Biochemistry, 42, (17) 4864-4873.
- Sabelko J, Erwin J, Gruebele M. (1998). Cold-denatured ensemble of Apomyoglobin: implications for the early steps of folding. Phys Chem B, 102, (10) 1806-1819.
- Lopez CF, Darst RK, Rossky PJ. (2008). Mechanistic elements of protein cold denaturation. J. Phys. Chem B, 112, (19) 5961-5967.
- Whitten ST, Kurtz AJ, Pometun MS, Wand AJ, Hilser VJ. (2006). Revealing the nature of the native state ensemble through cold denaturation. Biochemistry, 45, (34) 10163-10174.
- Simler R, Walsh G, Mattaliano RJ, Guziewicz NG, Perez-Ramirez B. (2008). Maximizing data collection and analysis during pre-formulation of biotherapeutic proteins. BioProcess International, 6 (10) 38-45.
- Agashe VR, Udgaonkar JB. (1995). Thermodynamics of denaturation of Barstar: Evidence for cold denaturation and evaluation of the interaction with guanidine hydrochloride. Biochemistry, 34 (10) 3286-3299.
- Neira JL, Gomez J. (2004). The conformational stability of the stretomyces coelicolor histidine-phosphocarrier protein. Characterization of cold denaturation and urea-protein interactions. J. Biochem, 271, (11) 2165-2181.
- Pace CN, Shaw KL. (2000). Linear extrapolation method of analyzing solvent denaturation curves. Proteins, Suppl 4, 1-7.
- Scholtz JM, Grimsley GR, Pace CN. Solvent denaturation of proteins and interpretations of the m value. Meth, Enzymol. 466. Academic Press, 2009. 549-565.
- Santoro MM, Bolen DW. (1988). Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl. alpha.-chymotrypsin using different denaturants. Biochemistry, 27(21), 8063-8068.)
- Lumry R, Eyring H. (1954) Conformational changes of proteins. Phys. Chem, 58,110-120.
Issue
Related topics
Biopharmaceuticals, Bioprocessing, Bioproduction, Formulation, Proteins, Research & Development (R&D)





